Molecular Genetics: Piecing it Together
Molecular genetics is the study of the agents that pass
information from generation to generation. These molecules, our genes,
are long polymers of deoxyribonucleic acid, or DNA. Just four chemical
building blocks--guanine (G), adenine (A), thymine (T) and cytosine (C)--are
placed in a unique order to code for all the genes in all living organisms.
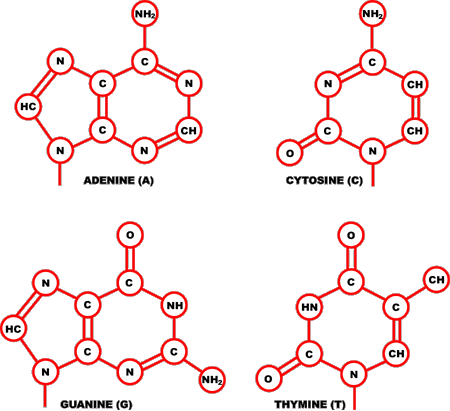
Figure 1. The Four DNA Bases
--------------------------------------------------------------------------------
Each deoxyribonucleotide acid (DNA) is made up of the sugar 2'-deoxyribose linked to a phosphate group and one of the four bases depicted above: adenine (top left), cytosine (top right), guanine (bottom left), and thymine (bottom right).
--------------------------------------------------------------------------------
Genes determine hereditary traits, such as the color of our hair or our eyes. They do this by providing instructions for how every activity in every cell of our body should be carried out. For example, a gene may tell a liver cell to remove excess cholesterol from our bloodstream. How does a gene do this? First, it will instruct the cell to make a particular protein. It is this protein that then carries out the actual work. In the case of excess blood cholesterol, it is the receptor proteins on the outside of a liver cell that bind to and remove cholesterol from the blood. The cholesterol molecules can then be transported into the cell, where they are further processed by other proteins.
Many diseases are caused by mutations, or changes in the DNA sequence of a gene. When the information coded for by a gene changes, the resulting protein may not function properly or may not even be made at all. In either case, the cells containing that genetic change may no longer perform as expected. We now know that it is mutations in the gene that codes for the cholesterol receptor protein that are associated with a disease called Familial Hypercholesterolemia. The cells of an individual with this disease end up having reduced receptor function and cannot remove a sufficient amount of low density lipoprotein (LDL), or bad cholesterol, from their bloodstream. A person may then develop dangerously high levels of cholesterol, putting them at increased risk for both heart attack and stroke.
How do scientists study and find these genetic mutations? They have available to them a variety of tools and technologies to compare a DNA sequence isolated from a healthy person to the same DNA sequence extracted from an afflicted person. Advanced computer technologies, combined with the explosion of genetic data currently being generated from the various whole genome sequencing projects, enable scientists to use these molecular genetic tools to diagnose disease and to design new drugs and therapies. Below is a review of some common laboratory methods that geneticists-- scientists who study the inheritance pattern of specific traits--can use to obtain and work with DNA, followed by a discussion of some applications.
Laboratory Tools and Techniques
The methods used by molecular geneticists to obtain and study DNA have been developed through keen observation and adaptation of the chemical reactions and biological processes that occur naturally in all cells. Many of the enzymes that copy DNA, make RNA from DNA, and synthesize proteins from an RNA template were first characterized in bacteria. These basic research results have become fundamental to our understanding of the function of human cells and have led to immense practical applications for studying a gene and its corresponding protein. For example, large-scale protein production now provides an inexpensive way to generate abundant quantities of certain therapeutic agents, such as insulin for the treatment of diabetes. As science advances, so do the number of tools available that are applicable to the study of molecular genetics.
Obtaining DNA for Laboratory Analysis
Isolating DNA from just a single cell provides a complete set of all a person's genes, that is, two copies of each gene. However, many laboratory techniques require that a researcher have access to hundreds of thousands of copies of a particular gene. One way to obtain this many copies is to isolate DNA from millions of cells grown artificially in the laboratory. Another method, called cloning, employs DNA manipulation procedures to produce multiple copies of a single gene or segment of DNA. The polymerase chain reaction (PCR) is a third method whereby a specific sequence within a double-stranded DNA is copied, or amplified. PCR amplification has become an indispensable tool in a great variety of applications.
Isolating DNA and mRNA from Cells
Cell Culture
Cell culture involves growing cells under artificial conditions, such as in the laboratory, either attached to some type of artificial surface or suspended in a special solution. In both cases, the cells are bathed in fluids containing nutrients that are either synthetically produced or extracted from related organisms. Certain cell types are more amenable to being grown in culture than others. For example, fibroblasts--a type of skin cell--have been cultured in the lab for decades, whereas the nuances of growing other cell types, such as nerve cells and stem cells, have only recently been elucidated. Conditions that serve to sustain one cell type may not apply to other cell types, or even the same cell type from another species. The conditions necessary for growing cells from humans, and many other mammals and plants upon which we depend, have been generally determined, whereas the conditions for culturing cells from exotic animals and plants still requires experimentation with each new species.
Cell culture is a useful technique because it provides a renewable source of cells for isolating DNA. In addition, scientists can use cells grown in culture to study how various chemicals and drugs affect certain cells, and by extrapolation, the whole organism. The process of growing cells outside a living organism, such as in a test tube, is referred to as in vitro. Once the effects of an agent on a cell have been thoroughly evaluated in vitro, the search for safe and effective treatments can be tested within a living organism, a process called in vivo testing.
DNA Isolation
DNA isolation refers to the process of extracting DNA from a cell in a relatively pure form. It involves separating DNA from other cellular components, such as proteins, RNA, and lipids. The cells used to obtain and isolate the DNA could come directly from tissue, or could be cultured laboratory cell lines obtained using the methods described earlier. Whatever the source, the DNA is isolated by placing the cells in a tube containing a special solution called a "cocktail,," and mechanically or chemically breaking them open. This causes the cell to release its contents into the cocktail containing enzymes, chemicals, and salts. Enzymes are used to chew up the proteins; chemicals to destroy any RNA present; and salts to help pull the DNA out of solution. At this point, the DNA will exist in long strands that form a mucous-like glob within solution. The DNA is then harvested by spinning the tube in a machine called a centrifuge. During spinning, the DNA collects in the bottom of the tube. The solution is then poured off, and the DNA is dissolved, or resuspended, in a second solution that will make it easy to work with in subsequent procedures. The result is a concentrated DNA sample containing many thousands of copies of each gene. For large-scale DNA analysis methods, such as those required to sequence the human genome, DNA isolation is performed using robots.
mRNA Isolation
Many researchers want to work with what is called expressed
DNA, or DNA that codes directly for the synthesis of a protein. This
special type of DNA is obtained by first isolating messenger RNA (mRNA)--an
intermediate between the expressed portions of DNA and the protein product.
Laboratory methods for mRNA isolation take advantage of a normal cellular
modification of mRNA--the addition of up to 200 adenine nucleotides to
one end of the mRNA molecule--called a poly-A tail. In the first step
of mRNA isolation, a cell is ruptured and the cellular contents are exposed
to synthetic beads coated with strings of thymine nucleotides.
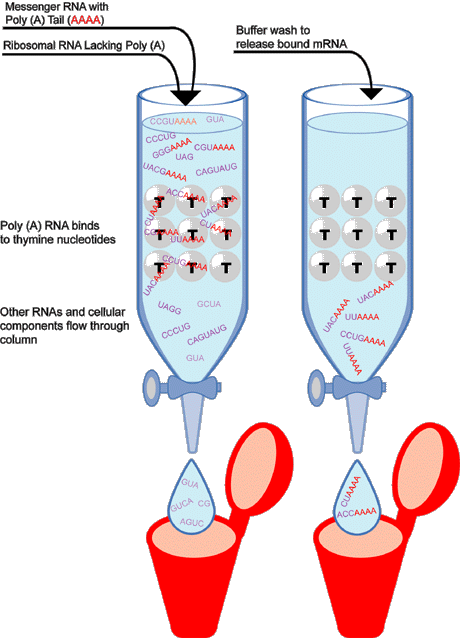
Figure 2. An Example of mRNA Isolation
--------------------------------------------------------------------------------
This cartoon demonstrates how Poly(A) RNA can be isolated from other RNAs by separation on a special solid support material. In this example, the material is made up of glass beads to which thymine molecules are attached. Since adenine and thymine molecules readily bind to each other, mRNAs with Poly(A) tails will be selectively retained on the beads. As seen on the left hand side of the diagram, a solution containing various RNA populations--including mRNAs with Poly(A) tails (designated red) as well as other RNAs and cellular material (designated purple)--is applied to the separation column. Only the Poly(A) RNA is retained, as it is immobilized on the solid support material. The other RNAs and cellular material pass through the column. On the right, the bound Poly(A) mRNA is retrieved by treating the column with a special buffer solution that breaks the thymine nucleotide-AAA bond. The mRNA can be collected in a tube for further experimentation.
--------------------------------------------------------------------------------
Since adenine and thymine readily bind to each other, Poly(A) mRNA is selectively retained on the beads while the other cellular components are washed away. Once isolated, purified mRNA is converted to single-stranded DNA using the enzyme reverse transcriptase, and is then made into a stable double-stranded DNA using the enzyme DNA polymerase. DNA produced in this way is called complementary DNA (cDNA) because it's sequence, at least the first strand, is complementary to that of the mRNA from which it was made. Why do researchers go to the trouble of making cDNA? cDNA is a much more stable compound than mRNA and, more importantly, because it was generated from a mRNA in which the non-coding regions have been removed, cDNA represents only expressed DNA sequence.
Methods for Amplifying DNA
Cloning DNA in Bacteria
The word "cloning" can be used in many ways. In this document, it refers to making multiple, exact copies of a particular sequence of DNA. To make a clone, a target DNA sequence is inserted into what is called a cloning vector. A cloning vector is a DNA molecule originating from a virus, plasmid, or the cell of a higher organism into which another DNA fragment of appropriate size can be integrated without interfering with the vector's capacity for self-replication. The target and vector DNA fragments are then ligated, or joined together, to create what is called a recombinant DNA molecule. Recombinant DNA molecules are usually introduced into Escherichia coli, or E. coli--a common laboratory strain of a bacterium--by transformation, the natural DNA uptake mechanism possessed by bacteria. Within the bacterium, the vector directs the multiplication of the recombinant DNA molecule, producing a number of identical copies. The vector replication process is such that only one recombinant DNA molecule can propagate within a single bacterium, so each resulting clone contains multiple copies of just one DNA insert. The DNA can then be isolated using the techniques described earlier.
A restriction enzyme is a protein that binds to a DNA molecule at a specific sequence and makes a double-stranded cut at, or near, that sequence. Restriction enzymes have specialized applications in various scientific techniques, such as manipulating DNA molecules during cloning. These enzymes can cut DNA in two different ways. Many make a simple double-stranded cut giving a sequence what are called blunt or flush ends. Others cut the two DNA strands at different positions, usually just a few nucleotides apart, such that the resulting DNA fragments have short single-stranded overhangs, called sticky or cohesive ends. By carefully choosing the appropriate restriction enzymes, a researcher can cut out a target DNA sequence, open up a cloning vector, and join the two DNA fragments to form a recombinant DNA molecule.
More on Cloning Vectors
In general, a bacterial genome consists of a single, circular chromosome. They can also contain much smaller extrachromosomal genetic elements, called plasmids, that are distinct from the normal bacterial genome and are nonessential for cell survival under normal conditions. Plasmids are capable of copying themselves independently of the chromosome and can easily move from one bacterium to another. In addition, some plasmids are capable of integrating into a host genome. This makes them an excellent vehicle, or vector, for shuttling target DNA into a bacterial host. By cutting both the target and plasmid DNA with the same restriction enzyme, complementary base pairs are formed on each DNA fragment. These fragments may then be joined together, creating a new circular plasmid that contains the target DNA. This recombinant plasmid is then coaxed into a bacterial host where it is copied, or replicated, as though it were a normal plasmid.
Bacterial plasmids were the first vectors used to transfer genetic information and are still used extensively. However, their use is sometimes limited by the amount of target DNA they can accept-approximately 15,000 bases, or 15Kb. With DNA sequences beyond this size, the efficiency of the vector decreases because it now has trouble entering the cell and replicating itself. However, other vectors have been discovered or created that can accept larger target DNA, including: bacteriophages--bacterial viruses that accept inserts up to 20 Kb; cosmids--recombinant plasmids with bacteriophage components that accept inserts up to 45 Kb; bacterial artificial chromosomes (BACs) that accept inserts up to 150 Kb; and yeast artificial chromosomes (YACs) that accept inserts up to 1000 Kb. Many viruses have also been modified for use as cloning vectors.
Polymerase Chain Reaction (PCR)
The polymerase chain reaction (PCR) is an amazingly simple technique that results in the eponential amplification of almost any region of a selected DNA molecule. It works in a way that is similar to DNA replication in nature. The primary materials, or reagents, used in PCR are:
- DNA nucleotides--the building blocks for the new DNA;
- Template DNA--the DNA sequence that you want to amplify;
- Primers--single-stranded DNAs between 20 and 50 nucleotides long that are complimentary to a short region on either side of the template DNA; and
- Taq polymerase--a heat stable enzyme that drives, or catalyzes, the synthesis of new DNA.
The PCR reaction is carried out by mixing together in a small test tube the template DNA, DNA nucleotides, primers, and Taq polymerase. First, the primers must anneal, or pair to the template DNA on either side of the region that is to be amplified, or copied. This means that the DNA sequences of these borders must be known so that the appropriate primers can be made. These oligonucleotides serve to initiate the synthesis of the new complementary strand of DNA. As taq polymerase--a form of DNA polymerase that catalyzes the synthesis of new DNA--is incredibly heat stable (thermostable), the reaction mixture can be heated to approximately 90 degrees centigrade without destroying the molecules enzymatic activity. At this temperature, the newly created DNA strands detach from the template DNA.
The reaction mixture is then cooled again, allowing more primers to anneal to the template DNA and also to the newly created DNA. The Taq polymerase can now carry out a second cycle of DNA synthesis. This cycle of heating, cooling and heating is repeated over and over. Since each cycle doubles the amount of template DNA in the previous cycle, one template DNA molecule rapidly becomes hundreds of thousands of molecules in just a couple of hours.
PCR has many applications in biology. It is used in DNA mapping, DNA sequencing, and molecular phylogenetics. A modified version of PCR can also be used to amplify DNA copies of specific RNA molecules. As PCR requires very little starting material, or template DNA, it is frequently used in forensic science and clinical diagnosis.
Preparing DNA for Experimental Analysis
Gel Electrophoresis: Separating DNA Molecules of Different Lengths
Gels are usually made from agarose--a chain of sugar molecules extracted from seaweed--or some other synthetic molecule. Purified agarose is generally purchased in a powdered form and is dissolved in boiling water. While the solution is still hot, it is poured into a special gel casting apparatus that contains three basic parts: a tray, a support, and a comb. The tray serves as the mold that will provide the shape and size for the gel. The support prevents the liquid agarose from leaking out of the mold during the solidification process. As the liquid agarose starts to cool, it undergoes what is known as polymerization. Rather than staying dissolved in the water, the sugar polymers crosslink with each other, causing the solution to "gel" into a semi-solid matrix much like "Jello", only more firm. The support also allows the polymerized gel to be removed from the mold without breaking. The job of the comb is to generate small "wells" into which a DNA sample will be loaded.
Once a gel has polymerized, it is lifted from the casting tray, placed into a running tank, and submerged in a special aqueous buffer, called a running buffer. The gel apparatus is then connected to a power supply via two plugs, or electrodes. Each plug leads to a thin wire at opposite ends of the tank. Because one electrode is positive and the other is negative, a strong electric current will flow through the tank when the power supply is turned on.
Next, DNA samples of interest are dissolved in a tiny volume of liquid containing a small amount of glycerol. Because glycerol has a density greater than water, it serves to weight down the sample and stops it from floating away once the sample has been loaded into a well. Also, as it is nice to be able to monitor a DNA sample as it migrates across a gel, charged molecules--called dyes--are also added to the sample buffer. These dyes are usually of two different colors and two different molecular weights, or sizes. One of the dyes is usually smaller than most, if not all, of the sample DNA fragments and will migrate faster than the smallest DNA sample. The other dye is usually large and will migrate with the larger DNA samples. It is assumed that most of the DNA fragments of interest will migrate somewhere in between these two dyes. So, when the small dye reaches the end of the gel, electrophoresis is usually stopped.
Once the gel has been prepared and loaded, the power supply is turned on. The electric current flowing through the gel causes the DNA fragments to migrate towards the bottom, or positively charged end of the gel. This is because DNA has an overall negative charge due the combination of molecules in its structure. Smaller fragments of DNA are less impeded by the crosslinks formed within the polymerized gel than are larger molecules. This means that smaller DNA fragments tend to move faster and farther in a given amount of time. The result is a streak, or gradient, of larger to smaller DNA pieces. In those instances where multiple copies of DNA all have the same length, a concentration of DNA occurs at that position in the gel, called a band. Bands can result from a restriction enzyme digest of a sample containing thousands of copies of plasmid DNA, or PCR amplification of a DNA sequence. The banded DNA is then detected by soaking the gel briefly in a solution containing a dye called ethidium bromide (EtBr). EtBr is an intercalating agent, which means that it is capable of wedging itself into the grooves of DNA, where it remains. The more base pairs present within a DNA fragment, the greater the number of groves available for EtBr to insert itself. EtBr also fluoresces under ultra violet (UV) light. Therefore, if a gel soaked in a solution containing EtBr is placed under a UV source, a researcher can actually detect DNA by visualizing where the EtBr fluoresces. Since a scientist always loads and runs a "control" sample that contains multiple fragments of DNA with known sizes, the sizes of the sample DNA fragments can be estimated by comparing the control and sample bands.
DNA Blotting
The porous and thin nature of a gel is ideal for separating DNA fragments using electrophoresis, but as we mentioned earlier, these gels are delicate and rarely usable for other techniques. For this reason, DNA that has been separated by electrophoresis is transferred from a gel to an easy to handle inert membrane, a process called blotting. The term "blotting" describes the overlaying of the membrane on the gel and the application of a pad to ensure even contact--without disturbing the positions of the DNA fragments. In the first step, the DNA trapped in the gel is denatured--the double-stranded DNA is broken into single strands by soaking the gel in an alkaline solution. This readies the DNA for hybridization with a probe--a piece of DNA that is complementary to the sequence under investigation. A membrane, usually made of a compound called nitrocellulose, is then placed on top of the gel and compressed with a heavy weight. The DNA is transferred from the gel to the membrane by simple capillary action. This procedure reproduces the exact pattern of DNA captured in the gel on the membrane. The membrane can then probed with a DNA marker to verify the presence of a target sequence.
Creating a DNA Library
In order to track the millions, or even billions, of nucleotides in the genome of an organism, scientists often create what is called a "DNA library," or a large collection of DNA fragments. There are many different kinds of libraries. A genomic library contains all the different types of DNA sequences found in a genome--introns, exons, non-coding, and repetitive DNA sequences. Scientists also make libraries exclusively of genes that are "expressed," or those genes that get transcribed into messenger RNA and then translated into protein. This library is called a complementary DNA (cDNA) library and is often made from mRNA expressed in a particular tissue type. A library is called chromosome-specific if the starting DNA came from just one chromosome.
Methods for Analyzing DNA
Once DNA has been isolated and purified, it can be further analyzed in a variety of ways, such as to identify the presence or absence of specific sequences or to locate nucleotide changes, called mutations, within a specific sequence.
Autoradiography: Probing DNA
To locate a specific DNA sequence, scientists rely on the base-pairing, or "hybridization," of a short piece of DNA that is complementary to the sequence of interest. This short, single stranded piece of DNA is called a "probe" and can be tagged with either mildly radioactive nucleotides or nucleotides that are linked to a substance that emits light when exposed to certain chemicals.
If we refer back to the blotting procedure described earlier, we mentioned that after the target DNA becomes trapped in the nylon membrane, the membrane is incubated in a solution that contains a probe. In this case, the probe would be radioactively labelled. Wherever the probe sequence complements a sequence on the membrane, it will anneal, or join together, to form a region of double-stranded of DNA. The membrane is then washed to remove all unbound probe and then exposed to a piece of x-ray film. The detection of radioactively labeled molecules by exposure to an X-ray sensitive photographic film is referred to as "autoradiography". Wherever the radioactively labeled probe has annealed to the test DNA, a black spot will be appear on the film.
This method is useful for a variety of applications. For example, suppose you know the DNA sequence of a particular gene (allele) that causes a disease. Now you want to know if a certain individual carries that allele. You can do this by following the steps outline above. First, isolate some of their DNA. Separate it out on a gel. Then, perform a Southern blot followed by autoradiography. If a black spot appears on the film, it indicates the presence of the disease-causing allele in that individual.
RFLP Analysis: Detecting Disease Genes
Every individual has slight differences or "sequence polymorphisms" that make their DNA sequence unique. These differences are often single base-pair changes that occur in regions of DNA that do not encode a gene, but which are recognized and bound by restriction enzymes. Restriction enzymes are protein that bind to a DNA molecule at a specific sequence and make a double-stranded cut at, or near, that sequence. Thus, when DNA from two different individuals is cut with a single restriction enzyme, DNA of different lengths will usually be produced. This is because not all restriction sites will exist within everyone's DNA. These variations in fragment length are referred to as restriction fragment length polymorphisms (RFLP), and the pattern of fragments is unique for each person. If a restriction polymorphism can be linked to a particular phenotype, such as eye color, it is called a restriction marker. Restriction markers are important because they offer a diagnostic procedure for detecting a disease and can help a researcher isolate a gene.
DNA Sequencing
The process of determining the order of the nucleotide bases along a DNA strand is called sequencing. In 1977, twenty-four years after the discovery of the structure of DNA, two separate methods for sequencing DNA were developed: the chain termination method and the chemical degradation method. Both methods were equally popular to begin with, but, for many reasons, the chain termination method is the method more commonly used today. This method is based on the principle that single-stranded DNA molecules that differ in length by just a single nucleotide can be separated from one another using polyacrylamide gel electrophoresis, described earlier.
The DNA to be sequenced, called the template DNA, is first prepared as a single-stranded DNA. Next, a short oligonucleotide is annealed, or joined, to the same position on each template strand. The oligonucleotide acts as a primer for the synthesis of a new DNA strand that will be complimentary to the template DNA. This technique requires that four nucleotide-specific reactions--one each for G, A, C, and T--be performed on four identical samples of DNA. The four sequencing reactions require the addition of all the components necessary to synthesize and label new DNA, including:
- A DNA template;
- A primer tagged with a mildly radioactive molecule or a light-emitting chemical;
- DNA polymerase--an enzyme that drives the synthesis of DNA;
- Four deoxynucleotides (G, A, C, T); and
- One dideoxynucleotide, either ddG, ddA, ddC, or ddT.
After the first deoxynucleotide is added to the growing complementary sequence, DNA polymerase moves along the template and continues to add base after base. The strand synthesis reaction continues until a dideoxynucleotide is added, blocking further elongation. This is because dideoxynucleotides are missing a special group of molecules, called a 3'-hydroxyl group, needed to form a connection with the next nucleotide. Only a small amount of a dideoxynucleotide is added to each reaction, allowing different reactions to proceed for various lengths of time, unti, by chance, DNA polymerase inserts a dideoxynucleotide , terminating the reaction. Therefore, the result is a set of new chains, all of different lengths.
To read the newly generated sequence, the four reactions are run side-by-side on a polyacrylamide sequencing gel. The family of molecules generated in the presence of ddATP are loaded into one lane of the gel and the other three families, generated with ddCTP, ddGTP, and ddTTP, are loaded into three adjacent lanes. After electrophoresis, the DNA sequence can be read directly from the positions of the bands in the gel.
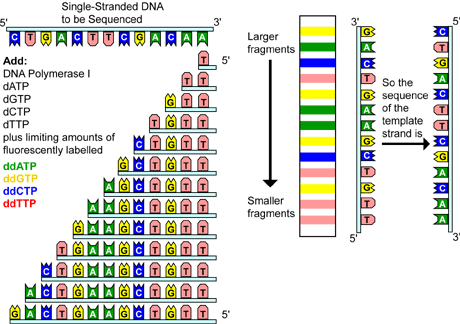
Figure 3. Chain Termination DNA Sequencing
--------------------------------------------------------------------------------
Chain termination sequencing involves the synthesis of new strands of DNA complementary to a single-stranded template (step I). The template DNA is supplied with a mixture of all four deoxynucleotides, four dideoxynucleotides--each labeled with a different color fluorescent tag, and DNA polymerase (step II). As all four deoxynucleotides are present, chain elongation proceeds until, by chance, DNA polymerase inserts a dideoxynucleotide. The result is a new set of DNA chains all of different lengths (step III). The fragments are then separated by size using gel electrophoresis (step IV). As each labeled DNA fragment passes a detector at the bottom of the gel, the color is recorded. The DNA sequence is then reconstructed from the pattern of colors representing each nucleotide sequence (step V).
--------------------------------------------------------------------------------
Variations of this method have been developed for automated sequencing machines. In one method, called cycle sequencing, the dideoxynucleotides--not the primers--are tagged with different colored fluorescent dyes, thus all four reactions occur in the same tube and are separated in the same lane on the gel. As each labeled DNA fragment passes a detector at the bottom of the gel, the color is recorded and the sequence is reconstructed from the pattern of colors representing each nucleotide in the sequence.
Chromosome Analysis
Cytogenetics is the field of science that deals with the relationship between human cells--and their chemical building blocks--and hereditary. Key to connecting chromosomes to symptoms and traits is the karyotype, a size-order alignment of chromosome pairs in a chart. The first such efforts to align the chromosome pairs, however, were quite crude. By 1959, about all that could be discerned was an extra or missing chromosome. Throughout the 1960s, pioneering cytogeneticists amassed techniques for capturing chromosomes at their most visible state. For most of a cell's existence, the chromosomal material is unwound and unable to absorb dyes. It is only during cell division that the chromosomes condense and become detectable. Researchers learned that treating cells with a hypotonic solution would cause them to swell, spreading apart the tangle of chromosomes. Another chemical agent, colchicine, was found to stop cell division when the chromosomes were at their most striking state. A third chemical, phytohemagglutinin, was found to entice lymphocytes--the blood cells most accessible for chromosomal study--to divide. With these tools in hand, the art of karyotyping was soon transformed into true science.
But still, chromosome pairs could not always be distinguished very well, and researchers had to rely on such large-scale and subjective clues as chromosome size and position of the centromere, a characteristically located constriction in each chromosome. Even staining the chromosomes distinguished unequivocally only four of the 23 chromosome pairs. These pairs were then grouped crudely by size and only large sections of extra or missing chromosomal material could be discerned.
By the 1970s, combining stains with digestive enzymes yielded far more subtle shading patterns, revealing the distinctive characteristic of each chromosome. Several different treatments were also developed that allowed researchers to further define the patterns of each chromosome. Now, tiny inversions--reversals in the banding pattern, duplications, deficiencies, and translocations--chromosomes that swap parts--could be detected. But building a karyotype required many hours of skilled work. The karyotyping procedure involved obtaining blood or some other appropriate tissue, separating out dividing cells, growing them in culturing, fixing them, and then dropping them onto a microscope slide. Then, using a light microscope, a researcher had to find a cell in which all of the untangled chromosomes were present and a photograph was taken. A print was then developed, and the individual chromosomes were cut out and arranged in pairs by size order into a chart, referred to as the karyotype. It is literally a scissors-and-tape operation and, believe it or not, many cytogenetics laboratories still depend chiefly on this method of chromosome analysis. But now an automatic chromosome analyzer--a system that includes a camera, a computer, and a microscope--may radically speed and improve the accuracy of the chromosome views.
Fluorescence In Situ Hybridization
Fluorescence in situ hybridization (FISH), a newer method for analyzing chromosomes, uses fluorescent molecules, called dyes, to "paint" genes on a chromosome. This technique is particularly useful for gene mapping and for detecting various chromosomal abnormalities. In this procedure, short sequences of DNA complementary to the sequence of interest--called probes--are hybridized to the sample DNA. Because the probes are labeled with fluorescent tags, a researcher can see the exact location of the DNA sequence of interest on a chromosome. An additional advantage of FISH is that it can be performed on nondividing cells, making it much more versatile than traditional karyotyping.
Scientists can actually create three types of FISH probes, each of which has a different application. Locus-specific probes hybridize to a particular region of a chromosome and are useful for detecting the location of a gene on a chromosome. Alphoid, or centromeric repeat probes are generated from repetitive sequences found at the centromeres of chromosomes. Because each chromosome can be painted a different color, researchers use these probes to determine whether an individual has the correct number of chromosomes. Whole chromosome probes are actually collections of smaller probes--called libraries--that each hybridize to a different sequence along the same chromosome. Using these libraries, researchers can paint an entire chromosome with various colors, generating what is called a spectral karyotpye. These types of probes are useful for examining both large- and small-scale chromosomal abnormalities.
Somatic Cell Hybridization
The term "somatic" cell refers to all the cells in an organism
that have differentiated into a specific cell type; excluding germ cells,
stem cells, and gametes. Somatic cell hybridization is the technique
of combining two cells from different tissues or species in a cell culture,
typically human and rodent, with the intent of deriving various cell lines
each with a different combination of chromosomes. The hybridized cells
fuse and coalesce, but their nuclei generally remain separate. However,
during cell division, a single spindle is formed so that each daughter
cell has a single nucleus containing sets of chromosomes from each parental
line. As hybrid cells grow and divide, they tend to randomly lose many
of their chromosomes until they reach a stable point. From there on out,
the cell will maintain the same number and species of chromosomes in subsequent
divisions. Little is known about the mechanisms behind this process, but
for some reason, hybrids between humans and rodents typically shed most
of the human chromosomes until only 8 to 12 of the original 46 human chromosomes
remain. Yet somehow, these cells can still survive. Through the careful
isolation and culture of different hybrid cell lines, researchers can
create a whole set of somatic cell hybrids which, together, contain the
entire complement of human chromosomes. Researchers can then use these
cell lines to screen for the presence or absence of a gene or gene product
(protein). For example, a researcher may test the cells ability to metabolize
a particular substance or study traits of antibiotic resistance. If a
cell line demonstrates an effect, the researcher can then study the chromosomes
present in that particular cell line to identify the gene that confers
the desired effect.
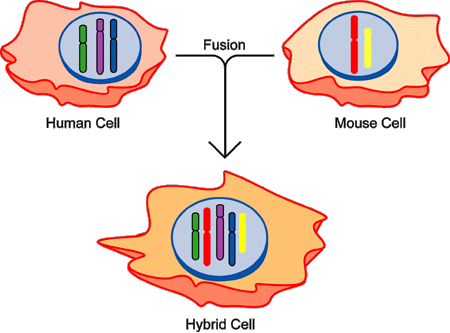
Figure 4. Somatic Cell Hybridization
--------------------------------------------------------------------------------
Human cells (left) and mouse cells (right), grown in culture, can be fused by treatment with a virus or chemical agent, yielding what is called a hybrid cell (bottom) containing both human and mouse chromosomes. In this example, three human chromosomes (depicted as green, purple and blue, with outlines) and two mouse chromosomes (designated red, and yellow, without outlines) are shown, with the hybrid cell having a mix of the five chromosomes.
--------------------------------------------------------------------------------
Impact of Molecular Genetics
Most sequencing and analysis technologies were developed from studies of nonhuman genomes, notably those of the bacterium Escherichia coli, the yeast Saccharomyces cerevisiae, the fruit fly Drosophila melanogaster, the roundworm Caenorhabditis elegans, and the laboratory mouse Mus musculus. These simpler systems provide excellent models for developing and testing the procedures needed for studying the much more complex human genome.
A large amount of genetic information has already been derived from these organisms, providing valuable data for the analysis of normal human gene regulation, genetic diseases, and evolutionary processes. For example, researchers have already identified single genes associated with a number of diseases, such as cystic fibrosis. As research progresses, investigators will also uncover the mechanisms for diseases caused by several genes or by single genes interacting with environmental factors. Genetic susceptibilities have been implicated in many major disabling and fatal diseases including heart disease, stroke, diabetes, and several kinds of cancer. The identification of these genes and their proteins will pave the way to more effective therapies and preventive measures. Investigators determining the underlying biology of genome organization and gene regulation will also begin to understand how humans develop, why this process sometimes goes awry, and what changes take place as people age.
![]()