G.Rothe1) and G.Schmitz1) for the Working Group on Flow Cytometry and Image Analysis. Members of the editorial committee: D.Adorf2), S.Barlafe1),Mgramatzki3),H.Hanenberg4),H.G.Höffkes5),G.Janossy6), R.Knüchel7),W.D.Ludwig8),T.Nebe9),C.Nerl2),A.Orfao10),S.Serke11), R.Sonnen12),A.Tichelli13),B.Wörmann14)
1) Institute for Clinical Chemistry, University Regensburg, Germany
2) Department of Hematology and Oncology, Städtisches Krankenhaus
München-Schwabing, Munich, Germany
3) III Medical Department, University of Erlangen, Germany
4) Department of Pediatric Hematology and Oncology, University of
Düsseldorf, Germany
5) Division of Hematology and Oncology, University of Magdeburg, Germany
6) Department of Clinical Immunology, Royal Free Hospital and School of
Medicine, London, UK
7) Institute for Pathology, University of Regensburg, Germany
8) Robert-Rössle-Clinic, Virchow Clinic, Humboldt University of Berlin,
Germany
9) Institute for Clinical Chemistry, Klinikum Mannheim, Germany
10) Department of Cytometry, University Hospital of Salamanca, Spain
11) Department of Hematology and Oncology, Universitätsklinikum
Rudolf Virchow, Free University of Berlin, Germany
12) St.George General Hospital, Hamburg, Germany
13) Department of Internal Medicine, University Hospital Basel, Switzerland
14) Department of Hematology and Oncology, University of Göttingen, Germany
Correspondence:
Prof. Dr. G. Schmitz
Institute for Clinical Chemistry and Laboratory Medicine
University of Regensburg
Franz-Josef-Strauß-Allee 11
D-93053 Regensburg, Germany
Tel: +49 (941) 944 6201, Fax: +49 (941) 944 6202
Abstract
Flow cytometry has become the preferred method for the lineage assignment and
maturational analysis of malignant cells in acute leukemias and lymphomas.
Multi-parametric immunophenotyping allows the detection of aberrant antigen
coexpression and the analysis of heterogeneity and clonality of malignant
cells in leukemias and lymphomas. The complexity of multiparameter analysis
techniques and the multitude of available monoclonal antibodies demand a
standardization of protocols for the use of flow cytometry in clinical
laboratories in order to achieve interlaboratory reproducibility. Therefore,
the Working Group on Flow Cytometry and Image Analysis has started an
initiative in order to establish a consensus protocol on the current
methods of the phenotyping of hematological neoplasias as a basis for
quality assurance and support for upcoming technologies such as quantitative
analysis of antigen densities and automated knowledge based analysis software.
In addition to general recommendations on assay procedures and quality control
specific recommendations are given for the selection of two-color reagent
panels and data interpretation in an attempt to define a basis for cross
evaluation against the different currently established laboratory protocols.
1. INTRODUCTION
Even before the availability of monoclonal antibodies (mAbs) against leukocyte differentiation antigens it was already apparent that immature blasts and lymphoma cells, despite morphological similarities, belong to different cell lineages and differentiation stages. Because of being a fast, objective, and quantitative method, flow cytometry has now become the preferred method for (1) lineage assignment, (2) maturational characterization of malignant cells, (3) detection of clonality, (4) heterogeneity and aberrant features of the malignant cell populations, and (5) quantitation of hematopoietic cells. The availability of fluorochromes with large Stokes shifts and automated closed flow cell cytometers as well as standard beads has increased reproducibility of the methods. Quantitative techniques regarding cell counts and antigen densities have been introduced into the analysis of immune deficiencies as well as hematological neoplasias. The use of knowledge based cluster software is a further step to improve flow cytometric analysis of hematopoietic malignancies in the clinical laboratory. In order to promote interlaboratory reproducibility of these methods, the goal of this consensus protocol is to guide the use of immunophenotyping techniques, selection of antibody combinations and fluorochromes, data analysis and internal quality assurance.
2. SCOPE OF FLOW CYTOMETRIC IMMUNOPHENOTYPING
The scope of flow cytometric immunophenotyping is to provide an objective and reproducible method for the diagnosis and therapy control of hematological malignancies, based on the immunological identification and characterization of clonally expanded abnormal precursor cells of the various hematopoietic lineages. This scope can be achieved in four different steps:
* Assignment of cellular lineage of the malignant cell
* Analysis of clonality, usually in conjunction with molecular and
cytogenetic findings
* Analysis of cellular maturation and heterogeneity within the malignant
cell populations
* Application of these observations in control and monitoring of therapy and
the detection of minimal residual disease
The final diagnostic classification of the disease is based on the immunological characterization of abnormal cells in conjunction with clinical findings and morphological and cytochemical analysis. These methods are particularly useful for the analysis of heterogeneous populations of malignant cells as well as therapy monitoring and detection of minimal residual disease.
2.1 Use of the analysis
The diagnostic value of flow cytometric immunophenotyping of bone marrow or periph- eral blood is established for the following hematopoietic malignancies: * Acute leukemias (lymphoblastic and myeloid) * Blast crisis in chronic myeloproliferative syndromes or leukemic transformation in myelodysplastic syndromes * Chronic lymphoproliferative disorders (T, B, and NK cell lineages) 2.2 Principle of the test The test is based on the identification of single or multiple surface membrane antigens or intracellular antigens of cells in suspension simultaneously with these cells' light scatter characteristics (Fig. 1, 2, Table 1-3). Non-nucleated cells are depleted before analysis by density gradient enrichment of mononuclear cells and/or erythrocyte lysis procedures. The antigens are detected with fluoresceinated mAbs, most frequently with direct immunofluorescence (IF). As a current standard this protocol suggests two-color IF analysis of antigens using fluorescein isothiocyanate (FITC) and R-phycoerythrin (R-PE) conjugated antibodies and either of three sample preparation techniques (Chapter 4.1). Table 4 and 5 give a list of antibody combinations which the authors recommend as a panel useful for a standardized analysis of hematological neoplasias. It is important to emphasize, however, that most flow cytometers used in routine work are equipped with a third photomultiplier in order to utilize three-color IF and only the quality of the third color reagent might be a limiting factor to introduce these more elegant methods (see below). The 3-color IF which may be currently used as an alternative technique will lead to a decrease of workload due to a lower number of tubes analyzed. Recently, also flow cytometric methods have been developed for the staining and analysis of intracellular antigens in suspension. These procedures allow the correlated multiparametric analysis of cellular antigens. Surface antigens can be discriminated from intracellular antigens if the staining of cells is compared before and after permeabilization. The pathological cell population is identified by a combination of uniform or abnormal light scatter characteristics, the relative expansion of cells of a single, often immature phenotype or an abnormal antigen coexpression. Abnormal scatter characteristics are based on the combination of forward scatter proportional to cell size and side scatter which is related to the granularity of cells. They help to correlate flow cytometric analysis to classical morphology. While the primary gating of samples being analyzed for forward and side scatter together with two- or three-color IF is still most frequently based on cellular scatter characteristics, a new trend is to apply primary immunological gating i.e. to focus on cells specifically stained by one of the antibodies first (e.g. precursor cell associated, T or B or myeloid specific) and then to analyze the other features of the immunologically identified populations at a second step of the analysis. The abnormal population identified based on scatter or immunological characteristics is then further characterized regarding cellular lineage, degree of maturation, abnormalities of antigen coexpression and heterogeneity in comparison to normal hematopoietic cells. A diagnosis is assigned in conjunction with clinical findings, morphology and cytochemistry based on the typical patterns of antigen expression in different clinically characterized disorders (Table 7-9).
Selection of antigens analyzed
A "minimal" primary panel may be used for lineage assignment of the
predominant blast population, followed by a secondary panel of mAbs
characterizing the definite phenotype and maturational stage of the blast
population depending on the results of the primary panel. This sequential
immunophenotyping of blasts is associated with some savings in reagent costs
but requires more time and planning. Alternatively, a predetermined panel
may be selected for an immediate direct, "extensive" characterization of a
wide range of immature and mature hematopoietic cells in the sample. This
"extensive" characterization may increase the sensitivity of the test
when malignant cells are only in a minority, and also help to characterize
the heterogeneity of pathological cells, as well as the concomitant
disturbances in the maturation of other cellular lineages. In addition, this
strategy offers a better control for consistency of staining results. This
"extensive" study in one step can be wasteful in reagents but saves time and
workload. Table 4 and 5 give a list of antibody combinations
which the authors consider useful for diagnostic discrimination of the below
discussed clinically defined entities. These combinations were selected
to maximize the discrimination of abnormal cells as well according
to the fluorochrome selection criteria discussed below. While other panel
combinations may be similarly useful the panels of Table 4 and 5
are suggested as a basis for the cross evaluation of different protocols.
Distinction of acute myeloid leukemia (AML) from acute lymphoblastic
leukemia (ALL). One of the most important applications is to define
the lineage derivation of acute leukemia. Lineage derivation can be precisely
determined by the analysis of functionally important cytoplasmic antigens
such as myeloperoxidase for myeloid cells, CD79 and CD22 for B-cell lineage
and CD3 for T-cell lineage (Table 1). Further membrane antigens
such as CD33 and CD13 for myeloid, CD19 for B-cell and CD7 for T-cell lineage
are also diagnostically helpful, but they are not always as precise as the
detection by cytoplasmic markers (Table 2). The additional analysis of
CDw65 and CD117 may increase the sensitivity for the detection of myeloid
cells. Analysis of multiple membrane antigens is mandatory as some of the
antigens expressed upon normal maturation may be lost during the leukemogenic
process (Table 2, 3).
Taken together, the detection of myeloperoxidase plus CD13/CD33 (myeloid), CD79 plus CD19 (B-cells) and CD3 plus CD7 (T-cells) respectively identifies >98% of acute leukemias as myeloid, B, or T and allocates 1.5-2% into the acute undifferentiated category. It is interesting that the irregularities in expression of certain membrane markers can paradoxically be helpful in leukemia diagnosis. For example, "lymphoid associated" antigens are expressed in cases of AML with known chromosomal aberrations as will be discussed below, e.g. AML with t(8;21) can be CD19+ and AML with t(15;17) can be CD2+. Nevertheless, these cases fail to express cytoplasmic CD79 or CD3 but are strongly MPO+. CD7 can also be detected, in addition to strong expression on T-ALL, in many immature cases of AML.
Classification of AML subtypes
Immunophenotyping also contributes to the further characterization of AML
subtypes, especially the M0 and M7 subtype, while the FAB-definition of M1
to M6 is still based mainly on morphology and cytochemistry. According to
recent knowledge, however, biologically defined subsets of AML such as the
t(8;21) karyotype with FAB M2 morphology, the inv(16) karyotype with FAB M4
morphology or the APL with t(15;17) express a characteristic immunophenotype
(Table 7). Whether the immunophenotyping may lead to a more
reproducible and clinically relevant characterization of all AML subtypes is
currently under investigation. The analysis of megakaryocytic antigens, e.g.
CD61, is mandatory as a secondary analysis in AML (Table 4). In
addition, it is suggested to include antigens characterizing neutrophils
(e.g. CD15), monocytes (e.g. CD14 or CD64), erythroid cells (e.g. glycophorin
CD2, CD19, CD56).
Classification of B-lineage ALL subtypes
The further goal in B-lineage ALL is the maturational analysis of B-cell
precursor ALL subtypes, i.e. pre-pre-B ALL (or pro-B-ALL), common B-ALL,
pre-B ALL and surface Ig-positive B-ALL. The secondary panel should include
the surface membrane markers CD22 and CD24 as additional B-cell associated
markers, as well as CD5 for the identification of subtypes of mature
B-lymphatic neoplasias (Table 4).
Classification of T-ALL subtypes
The goal is the discrimination of T-ALL and mature T-cell malignancies. The
secondary panel should include CD1a, CD2, CD5 as well as mAbs to T-cell
receptor (TcR) a/b and g/d chains. Furthermore the determination of CD4 and
CD8 and their coexpression may be helpful in the analysis of T cell
(im)maturity.
Chronic lymphoproliferative disorders
For the analysis of B-lineage non-Hodgkin's lymphoma it is suggested to use
mAbs or heterologous antisera against surface immunoglobulin light chains
(kappa and lambda) in combination with CD19 for the analysis of a clonal
expansion of B-cells (Table 5). The maturational characterization of
abnormal B-cells is then performed depending on the expression of further
subset specific antigens such as CD22, CD20, CD23, or FMC7, as well as
antigens that are typically coexpressed at certain stages of differentiation
such as CD5, CD10, or CD38. Other hairy cell leukemia associated antigens
include CD103 and CD11c (Table 8). The cellular analysis in chronic
leukemias of T-lymphocyte lineage is based on the expression of pan T-cell
associated antigens such as CD3, CD2 and CD5 together with antigens such as
CD4, CD8, CD56 and CD57 showing associations to subtypes of chronic leukemias
of T-lymphocyte lineage (Table 9). Further information can be gained
from the aberrant or missing expression of T-cell antigens because peripheral
T cell lymphomas variably show aberrations from the typical features of
peripheral T cells. These diagnostically important changes, unique to each
case, may include CD3 expression in aberrantly low density, lack of CD2 or
CD7, or negativity or positivity for both CD4 and CD8 expression.
3. MATERIALS
3.1 Test material
Types of specimenImmunophenotyping of nucleated hematopoietic cells can be performed using material taken from a variety of body compartments. Bone marrow is the preferred material for immunophenotyping in acute leukemia. Materials useful for immunophenotyping of hematopoietic cells include:
* Bone marrow
* Peripheral blood
* Liquor, or malignant effusions, e.g. ascites or pleural effusions
* Solid tissue, e.g. lymph nodes, after preparation of single cell suspensions
The minimum size of the pathological cell population in the sample which allows reproducible immunophenotyping is dependent on the analytical technique. One-parametric immunophenotyping is, by definition, unable to identify aberrant antigen coexpression except by comparing results of parallel tests, when large proportions of pathological cells in the mononuclear cell fraction uniformly express aberrant features. The sensitivity of multiparametric methods is dependent on the given abnormality and in some instances even minute subsets of cells can be detected. The sensitivity is increased when cells show highly abnormal light scatter characteristics.
Specimen collection
The date and time of specimen collection should be recorded. Specimens
should be transported to the flow cytometry laboratory as soon as possible. A
unique patient identification and information about age, sex, presumptive
diagnosis, differential blood count, current therapy as well as the status
of lymph nodes and spleen of the patient should be provided on the test
requisition. If it is likely that the material may not be analyzed within
6 hours of specimen collection, in the flow cytometry laboratory a white
blood cell, erythrocyte and platelet count should immediately be performed
from EDTA-anticoagulated material. This is also necessary if the
immunodiagnosis is performed in heparinized samples. In addition, smears
should be prepared from EDTA-anticoagulated material for morphological and
cytochemical analysis. If bone marrow is collected, white blood cell
differential counts should be performed simultaneously from bone marrow
and peripheral blood in order to check the degree of blood contamination
in the aspirate and the number of blasts in the blood. Polypropylene tubes
or syringes should be used for the collection and transport of the specimens
in order to avoid cellular adherence to polystyrene or uncoated glass.
Furthermore, collection systems with particulate ingredients, e.g. beads,
can damage cells, and should be avoided.
Anticoagulation
The most preferred anticoagulant, 50 U/ml preservative free heparin,
allows the analysis of Ficoll-Hypaque density separated cells for up to
three days after collection with a gradual loss of quality following a
longer delay. On the other hand, EDTA as anticoagulant has the advantages
that the losses of mature myeloid cells through adherence to the tube
are smaller and platelet aggregation may be reduced. Such platelet aggregates may
contaminate a scatter gate of small blast cells. An important advantage of
EDTA is that cells can be analyzed by morphology and automated hematology
analyzers using the same material. Nevertheless, cellular light scatter
characteristics deteriorate faster in EDTA than in heparin-treated samples.
Ficoll density gradient preparations of mononuclear cells prepared more
than 6 hours after specimen collection from EDTA-anticoagulated material
can be contaminated by neutrophils and red blood cells. Other anticoagulants
including acid citrate dextrose, solution A (ACD-A) may further prolong
cell viability. Nevertheless, currently only little experience is available
with these reagents.
Sample storage
Samples storage at room temperature (18 to 22 C) until analysis is recommended.
Specimens may be diluted (1:1) with adequately buffered filtered tissue culture media
which may also contain 1-2% fetal calf serum or bovine serum albumin. Storage at tem-
peratures below 10 C may lead to adsorption of immunoglobulins to cells and to a selec-
tive loss of cells or antigens. Certain cell types, e.g. Burkitt's lymphoma cells, however,
may not stable at higher temperature. Each laboratory should clarify how prolonged stor-
age at lower temperatures may alter membrane marker expression in the presence of the
given anticoagulants used in the laboratory under the particular storage conditions, lysing
and gradient separation procedures used in their routine analysis. Sample storage for >30
h should be avoided as the results can become irregular.
3.2 Reagents
Selection of clonesIt is now well documented that different clones of mAbs which recognize the same CD antigens may differ in their performance in the immunodiagnosis of leukemia. Some mAbs recognize epitopes sensitive to pH, or influenced by the presence of divalent cations, protease activity in the sample (e.g. in ascites) or which are dependent on the glycosyla- tion (e.g. CD15) or a genetic polymorphism of the antigen (e.g. CD16). Therefore, clones need to be selected carefully. Each laboratory should document in a "log book" the clones, fluorescence conjugates, and lots used for the analysis of a specific antigen. Preferentially, clones should be selected which have been characterized by the refer- ence laboratory and standardized according to clusters of differentiation (CD) by the International Human Leukocyte Typing Workshops. The reactivity of the antibody should be checked in comparison to published data.
For the intracellular detection of MPO in early myeloid blasts (myeloid leukemia without maturation, AML-M0) mAbs must be selected which have been shown to react not only with the active enzyme but also with the inactive proenzyme of MPO. The fixation and permeabilization procedures performed before incubation with mAbs in the analysis of intracellular antigens may lead to the selective destruction of epitopes important for the binding of mAbs. Therefore, mAbs used for the analysis of intracellular antigens must be tested for appropriate reactivity after the routine fixation used in the laboratory.
Selection of conjugates
The use of directly fluorochrome-conjugated mAbs in simultaneous two- or three-color
immunofluorescence is the preferred method for the analysis of cellular antigens. Some of
the currently available reference data have, however, been generated with indirect
staining techniques. These indirect methods and the direct staining techniques, which
use a range of fluorochromes, are associated with different thresholds for recognition
of antigen expression. For these reasons, the published results on weakly expressed
antigens from the different laboratories may not be readily comparable.
A given fluorochrome conjugation of a particular mAb should be chosen on the basis of density and heterogeneity of membrane expression of the antigen. Thus antigens with weak expression, such as CD13, CD19 and CD33 should be detected using mAbs conjugated to the bright fluorochrome R-phycoerythrin (PE) or the less bright Cy5 tan- dem conjugate (R-PE/Cy5), while strongly expressed antigens such as CD45 or Class II (HLA-DR) might be analysed with more dim conjugates such as FITC or PerCP. The combination of very bright and dim fluorescence on the two parallel channels can lead to artefacts due to the non-linearity of fluorescence compensation. In addition, the sterical hindrance in the simultaneous binding of different mAbs should be controlled when selecting combinations of mAbs.
Fluorochrome conjugations may also lead to non-specific binding of mAbs especially to myeloid cells. This may be the case with polyclonal antibodies conjugated with R-PE, e.g. anti-immunoglobulin antibodies, or conjugated with R-PE/Cy5 tandem fluorochromes. MAbs showing non-specific binding to myeloid cells, e.g. R-PE/Cy5 conjugates of CD19 binding to monocytes, should not be used for the immunophenotyping of hematological malignancies.
If directly fluorochrome-conjugated mAbs are not available for a specific antigen bio- tinylated mAbs may alternatively be used in combination with streptavidin-fluorochrome conjugates as secondary reagent that allow a more sensitive detection of cellular antigens then indirect IF staining. Furthermore, biotin-streptavidin labelling can be combined with directly conjugated mAbs for multiparameter assays. However biotin is present in the serum and in culture media and thus may lead to artefacts unless appropriate washing steps are performed.
MAbs should be stored according to the manufacturers description and repeated freezing and thawing should be avoided. Whenever a new lot of reagent is received, this should be titrated in comparison to the previous lot to ensure comparability of the staining results. Alternatively, an equal molarity of reagent may be determined using calibration beads with a defined binding capacity for mouse immunoglobulins. Buffers for antibody incubations and washing
Phosphate buffered saline (PBS) with sodium azide (0.1 to 0.2 %) and a protein solu- tion, e.g. bovine serum albumin (BSA), fetal calf serum (FCS) or human AB serum (0.1 to 2 %), is a typical buffer used during the incubation of the cell samples with mAbs and in subsequent washing steps. All protein containing media should be filtered to reduce "optical noise" presenting as "rare events". Azide reduces capping or internalization of membrane molecules after binding of the mAbs, which can occur during incubation above 10 C. The protein is added to the buffer in order to reduce nonspecific binding of the mAbs as well as loss of cells during washing procedures. Nonspecific binding is especially a problem during the multiple incubations in indirect staining. Losses of cells during washing steps depend on whether native or fixed cells are centrifuged, on the number and stringency of the centrifugation steps, centrifugation temperature, and on the types of tubes used (polystyrene or polypropylene). Careful control of these pitfalls may allow the use of PBS alone with the potential advantage of reduced optical noise during flow cyto- metric analysis.
Lysis reagents
An ammonium chloride solution (e.g. 8.29 g NH4Cl, 1.0 g KHCO3, 37 mg EDTA, pH
7.3), hypotonic sodium chloride solutions or a variety of commercial reagents may be used
for the lysis of erythrocytes. Most commercial lysing reagents contain a low concentration
of fixatives. To avoid destruction of epitopes or artificial staining of intracellular antigens,
these lysing solutions should only be used for erythrocyte lysis after the cells have been
loaded with mAbs. It is suggested to adhere to the manufacturers instructions when using
commercial lysing reagents.
Density gradients
Ficoll hypaque solutions (d=1.077) are recommended for the isolation of mononuclear
cells. Ready-to-use separation tubes containing membrane coated ficoll hypaque solu-
tions may be used to facilitate the separation procedure.
Fixants
Buffered solutions of formaldehyde (0.5 to 1 %) or paraformaldehyde (1 to 2 %) may be
used for the fixation of stained and washed cell preparations if the flow cytometric meas-
urement is delayed. The pH control of the solution is important as the fluorescence of
most fluorochromes is strongly pH-dependent.
3.3 Instrumentation
Flow cytometerA 488-nm argon laser based instrument is preferred with a closed flow cell. The instru- ment should allow the analysis of two or three fluorescences together with cellular forward and 90 light scatter both following logarithmic (at least 3.5 decades) or linear amplifica- tion. All detectors should be fine adjustable. List mode data should be storable on a removable mass storage device.
Software
The software for data acquisition should store the list mode data in a standardized for-
mat (e.g. FCS 2.0) together with a documentation of date and time of data collection, in-
strument parameter settings, as well as an operator-defined sample identification tag and
the list of reagents and fluorescence channels used in the test. The software for data
analysis should allow quantitative, linearized analysis of logarithmic data and logical
combinations of multiple analysis regions. All data manipulations during the analysis pro-
cedure, e.g. the definition of analysis regions and gates should be electronically storable.
4 METHODS
4.1 Sample preparation
Documentation of sample conditionsAbnormal sample conditions such as hemolysis, lipemia, partial draw in the case of ACD as the anticoagulant, and abnormal specimen temperature should be recorded. Specimens with visible clots or obviously frozen during transport as well as samples lacking a unique patient identification should be rejected.
Morphology / Cytochemistry
Morphological and cytochemical analyses of the specimens should be performed from
EDTA-anticoagulated material according to standard methods. If bone marrow suspen-
sions are sent together with bone marrow smears, a morphological cell differentiation
should be performed from the suspension as well as smears as these may be derived
from different fractions of the aspiration.
If material cannot be analysed by flow cytometry directly upon arrival, a morphological analysis and cell counts should be performed twice, i.e. upon receipt and before prepara- tion, to identify selective cell losses during storage. Similarly selective cell losses following density gradient separations may be observed.
Sample preparation
Erythrocytes may be depleted using three alternative methods.
(1) The material is first incubated with fluorochrome-conjugated mAbs followed by the
lysing of erythrocytes.
(2) Erythrocytes are lysed first and nucleated cells are incubated with mAbs.
(3) A suspension of mononuclear cells prepared by a density gradient centrifugation is
stained with the mAbs.
The methods using erythrocyte lysis are less time consuming. As with the exception of
some late normoblasts virtually no nucleated cells are lost the simultaneous characteriza-
tion of all cells including the more mature myeloid cells in the sample is possible. These
results can be quantitatively correlated to the morphological and cytochemical analysis of
the cells. Due to the higher complexity of the cellular populations, these methods have to
be used in combination with multiparametric immunofluorescence assays.
The simultaneous flow cytometric analysis of neutrophils after whole blood lysis proce- dures is of special interest in disorders of myeloid differentiation. Thus characteristic preleukemic alterations of neutrophils, such as a low side scatter signal corresponding to low granularity are detectable in myelodysplastic syndrome. Furthermore, in AML during complete remission as defined by hematological criteria, leukemia-associated alterations of mature myeloid cells may still be detectable as an indicator of clonal persistence. A potential drawback of this method is that when old specimens are analysed counterstaining of dead cells may be necessry to exclude non-specific binding of mAbs.
Density gradient separation of viable mononuclear cells leads to an enrichment of blast cells. For this reason in the past, when used with single colour analysis, the density gradient separation was preferred. An advantage of the method is that the prepared cells can be also optimally used for culture in vitro, for cryopreservation and molecular studies of leukaemic blasts. The density gradient separation may, however, lead to selective losses of pathological cells. For all separation methods, the recovery of cells should be documented.
Staining
The optimal technique is to utilize two- or three directly conjugated mAbs in tandem.
The sequential addition of different mAbs has also been sometimes recommended but
the additional cell losses due to the higher numbers of washing steps are a significant
shortcoming of this cumbersome method. Automated sample preparation are
preferable when compared to manual methods as possible handling errors are
reduced. These techniques frequently omit washing steps after incubation of cells with
mAbs ("no wash" procedures), particularly for the analysis of T cell subsets in HIV
seropositive individuals. Due to the higher background of unbound mAbs these
methods may not be suitable for the analysis of haemopoietic malignancies when the
antigen expression is weak.
Titration of mAbs
The concentration of all mAbs should be titrated towards a "saturating titer" when
the labelling optimally discriminates positive from negative populations. The optimal
concentration usually is the concentration double to that which results in a maximum
difference of fluorescence between positive and negative cells in the sample. Care
should be taken, however, when abnormal samples contain high numbers of cells
which express an antigen with particularly high density. In such cases mAb
concentration has to be increased.
Controls
Each staining procedure should include positive and negative controls. At least one
antigen should be selected as a positive control, which shows bright and homogeneous
expression on most cells of the sample, e.g. CD45 or HLA class I. Another control should
be selected which is expressed at different densities only on subsets of the cells. MAbs
against lactoferrin may serve as a positive control in the intracellular analysis of antigens
resulting in the staining of contaminant mature myeloid cells in the sample.
As a negative control cellular autofluorescence as well as the binding of nonspecific
mAbs of the same isotypes conjugated to the same fluorochromes should be recorded.
Directly fluorochrome conjugated isotype controls from different manufacturers, however,
significantly differ in their staining characteristics due to different fluorochrome to protein
ratios as well as different conjugation procedures. Isotype controls, therefore, are only of
limited value when using multiple directly conjugated mAbs from different manufacturers.
Alternatively for specific antibodies the amount of nonspecific binding may be determined
based on the fluorescence signal of reportedly negative normal cells in a sample (e.g.
CD3 on neutrophils and monocytes).
Biological or other type of standards may be used in addition as external controls in the test system.
Fixation
Samples can be fixed using buffered solutions of formaldehyde or paraformaldehyde if
not analyzed immediately. These samples are stable for several days. The low amount of
fixatives in commercial lysing solutions is not adequate for a prolonged storage. Fixation
may be especially necessary following the use of a non-fixative erythrocyte lysing solution
which may lead to kinetically instable light scatter characteristics of the nucleated cells.
Procedure for the counterstaining of dead cells.
Old samples or samples prepared by disaggregation of solid tissue may contain dead cells. When simultaneously counterstained with propidium iodide dead cells can be iden- tified and excluded during analysis. Less well excluded dyes such as 7-amino-actinomycin D may be better suited for multi-parameter staining. Viability also may be assessed by light microscopy using trypan blue. These methods are, however, only appropriate for samples that have not been fixed or permeabilized.
Erythrocyte lysis techniques with fixation should not be performed on cell samples containing non-viable cells. Bone marrow, peripheral blood or liquor samples containing a significant amount of dead cells, e.g. 20 %, should be excluded from analysis due to the potential selective loss of abnormal cell populations unless no repeated sample can be obtained and an abnormal population of cells can be unambigously identified.
4.2 Measurement
Instrument alignment and data acquisitionFluorescence and scatter signals of 10-20x103 nucleated cells per sample should be acquired in list mode. Higher numbers of cells, 50x103, are required for the analysis of minimal residual disease. Logarithmic amplification and at least 256 channels resolution are required for immunofluorescence signals. Ideally the logarithmic amplification of light scatter signals is preferable for the analysis of bone marrow compared to linear amplifica- tion, as large cells, e.g. plasmocytoma cells are not analyzed "off scale". However, most instruments on the market do not allow logarithmic scatter amplification with a low dyna- mic range (e.g. 2 decades). Thus linear amplification of the light scatter signals may require different gain settings for an optimal resolution for some aberrant cell types or following different sample preparation protocols e.g. for surface and intracellular antigens. Either the forward or 90 -light scatter signals should be used as the threshold parame- ters depending on which gives the best discrimination between nucleated cells and debris. Thresholds or "gates" should be defined for the instrument using a normal blood sample. These boundaries should include all nucleated cells in the sample. Backgating on the cellular DNA following counterstaining with PI for fixed cells or LDS-751 in non-fixed cell samples may help in the definition of this threshold. When samples from treated patients are studied additional "live gates" may be defined based on known patterns of abnormal antigen coexpression for the cellsubset of interest, i.e. the blast populations which may still be present at low frequency.
The setup should be optimized to allow the analysis of multiple antigens at a low den- sity. A photomultiplier (PMT) voltage should be selected for each parameter, with a visible weak signal for background fluorescence on unstained cells. The fluorescence compen- sation should be based on a set of calibration beads or cells strongly stained with the same dyes used for immunofluorescence (e.g. FITC, R-PE, R-PE/Cy5 or PerCP). Auto- mated software-controlled fluorescence compensation is preferable. In a second step, cells stained with two- or three-parameter combinations of CD3, CD4, and CD8 may be used to control this compensation. Software compensation of list mode data acquired withour compensation is an alternative approach for the elimination of fluorescence over- lap.
Calibration
The optical sensitivity and linearity of the instrument should be adequatly controlled. As
a general rule, the instrument should be calibrated daily using stable control material, e.g.
beads with different defined amounts of fluorochrome. Log sheets should be kept for PMT
settings and the respective mean and CVs of the fluorescence and light scatter signals on
stained and nonfluorescent beads.
Waste
Sodium hypochlorite (e.g. 1/10 of a 0.71 M sodium hypochlorite solution) should be
added to the waste container in order to inactivate biologically active material.
5 QUALITY ASSURANCE AND TROUBLE SHOOTING
5.1 Quality assurance
Quality assurance is based on three sets of observations: (a) the phenotyping of
normal samples as reference material, (b) internal control of results based on
consistency checks, and (c) on external controls performed in comparison with other
methods. Reagents and instruments should be regularly tested and documented in a
log book with these three systems. Laboratories should occasionally analyse replicate
blood and bone marrow samples from normal donors and patients in order to identify
the precision of their analysis.
"Internal" consistency checks
Consistency of staining characteristics on normal cells
As an internal quality control, the binding of various mAbs to normal cells should be
known. Obviously, the percentages of normal cells expressing antigens with similar
distribution should be compared. For example, the pan-T reagents CD2, CD3, CD5
and CD7 should show a similar, although not identical, distribution on bona fide T cells.
For B cells the numbers expressing B-cell associated antigens CD19, CD20, CD37
and CD22 should be similar, while CD23 and FMC7 are expected to react with a
smaller subset. Furthermore, for cells with monocyte scatter the numbers of CD33bright
cells should be similar to the CD14++ and CD4+/CD3- cells. For neutrophils, the
expression of CD15 should be similar to the numbers of CDw65bright cells. Finally, for
erythroid cells the percentage of CD45-negative, glycophorin A-positive cells should also
be similar. These consistency checks in normal peripheral blood should allow a com-
parison of the sensitivity and specificity of different reagents used.
Consistency of check sums
As a further control check sums may be performed. The sum of CD4+/CD3+ and
CD8+/CD3+ cells should be close to the total of CD3+ T lymphocytes. The sum of kappa-
light chain expressing B cells (e.g. CD19+ cells) and lambda+/CD19+ B cells should cor-
respond to the total amount of CD19+ B-cells in normal peripheral blood. The sum of T
cells, B cells, and NK cells should be similar to the total amount of lymphocytes deter-
mined by gating on CD45+/CD14- expression.
"External" consistency checks
As an external check the quantitative or qualitative cellular phenotypes obtained by
immunophenotyping should be compared to the results of the morphological and cyto-
chemical differentiation of cells identified on automated hematology analyzers. Further-
more, the characteristics of cellular staining with specific antibodies, especially against
intracellular antigens, should be regularly controlled by fluorescence microscopy.
5.2 Trouble shooting
Problems associated with sample preparationCell losses during washing
(Selective) losses of cells may occur during multiple washing steps. Possible solutions for the problem are a reduction of washing steps, washing at 4 C instead of room tem- perature, increasing the amount of protein added to the washing buffer, the addition of a Ca++-chelator such as EDTA to the sample, or the use of tubes with non-adhesive sur- faces, e.g. polypropylene. Also a non-fixative erythrocyte lysing solution may be associ- ated with a reduced loss of cells during further washing steps.
Incomplete lysis or high amounts of debris
Inadequate lysis of erythrocytes may occur upon preparation of lipidemic blood samples
following inadequate agitation during lysis, as a result of an increased amount of
reticulocytes in cryoglobulinemia or with samples obtained after intensive chemotherapy.
The solutions of the problem are DNA staining followed by triggering on nucleated cells,
washing of cells before staining, or cells may be prepared by density gradient separation.
Nonspecific staining
Staining of cells with multiple mAbs may be due to a population of nonviable cells. This
can be checked with propidium iodide in unfixed samples. A high nonspecific background
of fluorescence may also be due to increased autofluorescence of the cells, increased Fc-
receptor binding of antibody, or inadequate washing procedures. A high autofluorescence
may occur in hypergranular promyelocytes, macrophages, erythropoietic precursors,
vacuolated blast cells, following drug treatment or as a consequence of prolonged storage
of cells following fixation. Autofluorescence can be reduced by selecting a fluorochrome
which spectrally emits at a different wavelength or by quenching of autofluorescence with
crystal violet. Nonspecific binding of mAbs to Fc receptors may be reduced by a preincu-
bation of cells with a high amount of nonspecific immunoglobulin before incubation with
the specific fluorochrome-conjugated mAbs. A less efficient method is the use of a higher
amount of human or bovine protein as a buffer constituent during the incubation steps
(see 3.1: Buffers). Nonspecific binding of antiplatelet mAbs to leukocytes or leukemic cells
may depend on the adhesion of platelets to monocytes and could lead to a misdiagnosis
of "CD61+ megakaryocytic leukemia if the investigator does not exclude this artefact. This
phenomenon may be reduced by carefully washing the cells before incubation with the
mAbs.
Problems associated with flow cytometric measurement
A high amount of debris can lead to difficulties in defining a scatter gate for nucleated
cells. Cellular DNA may be counterstained, e.g. with LDS-751 for viable cells or with
propidium iodide for fixed cells, to visualize the nucleated cell fraction.
A significant overlap of fluorescence may occur despite adequate compensation if cel-
lular antigens expressed at high densities are analyzed simultaneously with other antigens
that are weakly expressed. This may be due to irregularities of the electronic amplification
characteristics, due to an "inner filter" effect which occurs at high local concentration of
fluorochromes, or due to the dissociation of tandem fluorochromes.
Problems associated with data analysis
No clear separation of blast populations in bone marrow
Increased numbers of normal hemopoietic precursor cells, e. g. in regenerating bone
marrow, may mask the presence of small populations of abnormal cells. The precise
determination of leukemia associated phenotypes at presentation of the disease, often
characteristic to the malignant blast cells of an individual patient, is an essential require-
ment. The knowledge of these features together with the availability of stored list mode
files then facilitates the careful re-investigation of the sample that has been taken following
remission induction therapy. The determination of the abnormal phenotype from peripheral
blood may be helpful in the characterization of small blast cell populations.
Low expression of antigens
Cellular antigens may be expressed at an only low density with no clear separation of
positive and negative populations. This may be due to a low signal of the antibody used or
a wide heterogeneity of antigen expression within the population. A brighter conjugate,
e.g. PE, or R-PE instead of FITC should be used. Heterogenous populations may be
resolved by multiparametric counterstaining with additional antigens.
6 EVALUATION
The evaluation of the flow cytometric data is a stepwise procedure for the presentation of the clinically relevant information obtained from the flow cytometric list mode data:(1) List mode data are analyzed in a standardized manner in order to group cell popula- tions on the basis of density and homogeneity of antigen expression and on the cells light scatter features.
(2) Patterns of antigen expression are related to phenotypes of normal or abnormal hematopoietic differentiation.
(3) Data are ranked specifically related to disease and clinically interpreted. The first two steps of the data evaluation represent a technical analysis related to ref- erence data generated by flow cytometric analysis of normal bone marrow whereas the data interpretation is based on the integrated analysis of the immunophenotyping, other laboratory tests and clinical findings.
6.1 Data analysis
Analysis of antigen expression patterns on all cells In a first step, the antigen expression should be analyzed on all cells without a selection of analysis regions. Two-dimensional dot plots should be generated for antigen coex- pression patterns as well as for correlated analysis of antigens and light scatter signals. This step is necessary for both the detection of small abnormal populations based on their atypical antigen expression and for the identification of normal cellular components in the sample. These normal cells provide sufficient data for a rapid conclusion about the satis- factory consistency of results, i.e. about the high quality of the staining. Furthermore, some populations become visible on a two-parametric immunofluorescence dot plot which would be off-scale in light scatter analysis (e.g. plasmocytoma or large blasts). Popula- tions identified by their abnormal features may then be gated for further study. Population-specific analysis of antigen expression.The goal of the further analysis is the identification and further characterization of abnormal cell populations. The direct identification of an abnormal cell population in un- gated two-parametric presentations of multi-parametric list mode data especially when using erythrocyte lysis procedures is highly dependend on individual expertise and difficult to standardize. Therefore, the division of cells into analysis regions based on their light scatter allowing a correlated analysis of antigens detected from different tubes for each of the populations is suggested as a standardized method to reduce the complexity of the data. An assortment of cells into multiple light scatter-coded regions is based on the identification of normal lymphocytes, monocytes and neutrophils by CD45 and CD14 expression (Fig. 3). The corresponding light scatter regions are then identified based on a backgating of the populations characterized by their specific antigen expression. The three regions are (1) lymphocytes and immature blasts, (2) monocytes and more differentiated hematopoietic precursor cells, and (3) neutrophils and other mature myeloid elements. If no normal population is found within the sample, regions should be defined according to "standard" settings. The antigen expression is then separately analyzed for one or more of these scatter coded regions. The correlation between the specific antigen expression and scatter profile is also helpful for interpreting the results. CD4+/CD3- cells with a scatter profile of lymphocytes and immature blasts correspond to myeloid progenitor cells, whereas the same immunophenotype with a scatter profile of CD14+ cells is indicative of differentiated monocytes.
Abnormal cells may be found outside of the scatter-coded regions, and their percent- ages should be controlled on the basis of analysis of antigens expressed on all cells, e.g. the sum of the CD45 plus glycophorin A expression.
Definition of analysis regions based on backgating on abnormal cells
The exact phenotype of abnormal cells may be more reliably defined based on the
definition of the light scatter region which defines abnormal cells. This region may be
based on the backgating of cells showing abnormal patterns or expression of early pre-
cursor cell associated antigens either directly from ungated list-mode data or following the
above described scatter gating procedure. Abnormal cells are identified on their light
scatter characteristics alone when they are dominant in high count leukemia. The antigen
expression is then rapidly characterized and establishes the lineage derivation (see above
and Table 1).
Multi-dimensional cluster analysis as a tool for the direct multiparametric identification of normal cellular phenotypes as well as typical or previously defined blast phenotypes is a new concept which probably will significantly increase the precision of data analysis. However, the limited availability of software products until now does not allow a stan- dardization of this approach.
Methods for the analysis of antigen expression The antigen expression by light scatter-coded populations of cells can be analyzed at different degrees of complexity dependent on the multiparametric analysis. The heteroge- neity of antigen coexpression on abnormal populations and the analysis of relative densi- ties of antigen expression can be related to differentiation.
Characterization of predominantly expressed antigens
The simplest approach for analysing antigen expression in a light scatter defined
population is based on the definition of staining above threshold level. This level is directly
above the non-specific staining of cells which fail to express the antigen. This nonspecific
level of staining is determined by using nonspecific control mAbs and cells which are
negative for a certain antigen. Different populations of cells characteristically show
different levels of autofluorescence as well as different levels of nonspecific binding of
mAbs. The percentage of cells with an antigen expression above threshold is then
determined for all cells within the analysis gate.
When multiparametric data are evaluated analysis extends to the coexpression of dif- ferent antigens. For each of the fluorochromes a threshold is defined. This provides three positive regions for two-colour (1: F1+/F2-, 2: F1-/F2+, 3: F1+/F2+) and eight positive regions for three-colour immunofluorescence (Fig.4).
Analysis of antigen density
The complexity of data evaluation is increased when the antigen density is quantita-
tively analyzed on a population of abnormal leukemic cells in comparison to normal
populations. The relative expression density of myeloid markers such as CD14, CD33 or
CDw65 in addition to the cellular light scatter characteristics can be used to define normal
development along the granulocytic and monocytic pathways. Abnormal cell populations
when compared to normal cells, often reveal gross disorders of decreased or increased
expression within the malignant clone. The calibrated measurement of surface antigens
using reference beads coated with murine Ig (M-Ig) or with anti-M-Ig may help to establish
objective criteria for this quantitative approach but the details are beyond the scope of this
review.
6.2 Data presentation
For the laboratory, the technical parameters which may affect the quality of the analysis have to be reported. For the physician, the immunophenotype of the cell populations should be reported in comparison to phenotypes of normal cells during hematopoietic differentiaton. The assays performed should be given in order to allow comparison of data between different laboratories.
Laboratory report form
A standardized laboratory report form should document the time when the sample was
received, prepared and analyzed. If the analysis was not performed within 6 h of specimen
collection, the viability of cells should be determined. The test parameters and the
instruments used for measurement should be visible both from the report forms and log-
books maintained for reagents and flow cytometers. Furthermore, the name and long-term
storage of the list mode files and other files that document the analysis (gate and region
information) should be recorded. Print-outs of signals of ungated cells as well as the gates
and regions defined for the analysis might be attached to the laboratory report form to
remain accessible if queries arise when the presence of leukemic cells is re-investigated
following therapy (see below).
The name of the operator and the decisions made during the handling of the sample should be documented. This includes the reagent panel used, based on the morphological analysis and the physician request.
Physician report form
The physician report form should document the request by the physician, type of mate-
rial received and time of receipt. Unfavorable conditions that may influence the interpreta-
tion of data, e.g. reduced viability, clots, or a partially frozen sample, should be noted.
Also, results that remain suspect due to unresolved problems (related to sample prepara-
tions, unexpected irregularities in reagent performance or instrument reliability) should be
noted on physician report form.
The minimal result should contain the concentration of nucleated cells in the sample, a
brief description of normal cellular elements based on morphology and/or immunopheno-
typing, the percentage of abnormal cells in the sample and criteria for their identification
together with the expression of the most important antigens on the abnormal population.
In addition, the heterogeneity of the abnormal population and the size and specific pheno-
types of the main subpopulations of the abnormal cells should be explained.
As a help for the interpretation of the test the report form should describe the immuno-
phenotype of normal cells during hematopoietic differentiation when analyzed with the
same assay system. Preferably, reference values should be given for the size of the cor-
responding main normal populations, in order to clearly explain the nature and the degree
of disorder represented by the aberrant cells seen in the pathological sample.
An abnormal population of cells may be identified by several criteria:
- Expansion of cells of a single lineage particularly if it is accompanied by a block
of maturation and accumulation of immature cells
- Clonality of a population (selective expression of kappa or lambda light chains)
- Asynchronous expression of antigens, e.g. TdT on sIg+ B cells or CD34 on
CDw65 positive cells
- Decreased or increased expression density of a differentiation antigen
- Increased phenotypic heterogeneity of cells with similar scatter characteristics
- Abnormal coexpression of antigens associated with different lineages
- Abnormal maturation or reduction of mature elements of the same lineage
Lineage assignment and further characterization of blast population in acute
leukemias
Lineage assignment
Immature blast cells may express at the same time markers associated with different
lineages. In addition, different subpopulations of abnormal cells may express antigens not
related to each other. The relative assessment of the role of different of these antigens in
the decision about the cellular lineages may depend on the number and degree of lineage
association of these antigens as well as on the density and heterogeneity of the cellular
expression of these antigens. A further criterion may be the presence of an antigen on a
minor subset of the predominant population. Furthermore, the expression of an antigen at
a relatively lower density compared to the physiological expression at the corresponding
phenotype of differentiating hematopoietic cells, a high heterogeneity of expression den-
sity, or expression only on a subset of the abnormal population may be indicators of an
aberrant coexpression of an antigen. A consensus on such criteria for the assignment of
the cellular lineage in the case of aberrant antigen coexpression or a heterogeneity of the
blast cell populations has just been suggested by Bene et al. (Leukemia 9: 1783-6, 1995)
in an initiative of European reference laboratories (EGIL classification).
Maturational analysis
The further characterization of the blast cell population regarding the maturational stage
of the population is the second parameter of interest for the classification of the disease.
Also here a heterogeneity of the population or the asynchronous coexpression of antigens
may be major problems in the correct determination of the cellular phenotype. As a current
consensus it is recommended to report the maturational phenotype of the most
predominant population of blast cells.
Further characterization of blast cells
Specific cellular abnormalities of the blast cell populations may be of interest both for
the recognition of prognostically unfavorable subgroups of patients as well as for the
analysis of abnormal cells following therapy. Such abnormalities are the patterns of anti-
gen coexpression on different subpopulations or the light scatter characteristics of the ab-
normal population.
Characterization of normal cells
Additional disease specific information may be obtained from the analysis of abnor-
malities in the differentiation of more mature cells in the samples. Also a concomitant acti-
vation or expansion of immunologically relevant mature cells should be analyzed.
Correlation to morphology
Initially, or in parallel with the immunophenotyping, a morphological and cytochemical
classification of the blast cells should be performed according to the FAB classification.
Typical antigenic profiles in acute leukemia
As immunophenotyping is currently performed using different methods which often lead
to complex cellular phenotypes no general flow chart can be given for the classification of
different subgroups of acute leukemias. Most authors agree on the following phenotypes:
AML
As a general criterion for the classification as AML, except the subforms AML-M6
(erythroleukemia) and AML-M7 (megakaryoblastic leukemia) and sometimes AML-M5,
MPO should be demonstrated in the abnormal cell population either cytochemically or
immunologically. Furthermore, the diagnosis of AML is supported by the expression of
lineage-associated markers such as CD13, CD33, or CDw65. In some cases of AML
expression of antigens such as CD13 may be found only cytoplasmic rather than on the
cell membrane. The expression of CD34 or CD7 which is frequently found in AML is con-
sidered as a marker related to blast cell immaturity rather than a hint for cellular lineage.
Other markers are less frequently (CD2) or very rarely (CD10, CD19, and CD20) coex-
pressed. The coexpression of these markers, however, should not fulfil the criteria for a
biphenotypic leukemia (see below).
A specific expression of more mature myeloid markers or specific patterns of aberrant antigen expression are associated with different subclasses of AML (Table 7). The defini- tion of AML-M0 is based on the detection of MPO immunologically or by electron micros- copy in the absence of a cytochemical reaction of the inactive proenzyme. In addition, CD13 and / or CD33 and other myeloid markers should be expressed in the absence of lymphoid markers except for CD7 or TdT. The hypergranular promyelocytic leukemia AML-M3 (t(15;17) is characterized by the absence of HLA-DR. The myelomonocytic leu- kemia AML-M4 and the monocytic leukemia AML-M5, when compared to other forms of AML, often show simultaneous expression of CD14 and CD64. The expression of CD4 on blast cells is not restricted to AML with a monocytic component. AML-M6 and AML-M7 are characterized by the expression of antigens associated with erythroid or megakaryocytic antigens occasionally in the absence of panmyeloid antigens.
ALL
TdT is no specific marker for ALL as the marker is also found in 20 % of AML. In most
cases of ALL, however, the blast cell population shows expression of TdT, although TdT is
not found in the rare most mature subtypes of B-lineage ALL (i.e. B-ALL) or in some cases
B-lineage ALL
An ALL of the B-lymphocyte lineage is assumed if CD22 or CD79a expression is found
either cytoplasmic or on the cell surface with the expression of CD19 and HLA-DR. The
simultaneous expression of a single or two myeloid antigens can be found in a subset of
ALLs of the B-lineage. The expression should, however, not fulfil the criteria for the diag-
nosis of a biphenotypic leukemia as given below. Furthermore, a dim expression of CD22
may be a specific feature of some cases of acute myeloid leukemia.
T-lineage ALL
An ALL of the T-lymphocyte lineage is assumed if CD3 expression is found either
cytoplasmically or on the cell surface with the simultaneous expression of CD7. T-lineage
ALL subtypes can be defined based on the surface expression of CD1a, CD2, CD3, CD4
and CD8.
Biphenotypic acute leukemia
A biphenotypic acute leukemia may be found in two different forms. The abnormal cells
may show expression of markers from more than one lineage simultaneously or, in the
more rare cases, two distinct populations of blast cells are found which differ in lineage. A
single or multiple unexpected markers are increasingly found in acute leukemias depend-
ent on the increase in dimensionality and sensitivity of the analysis methods. The degree
of lineage association of specific antigens and the degree of coexpression of an antigen
on the majority of blast cells or only on a significant subpopulation (more than 10 %) are
helpful parameters for the classification of aberrant combinations of antigens. The Euro-
pean group for the immunological characterisation of leukemias (EGIL) established a
scoring system to define cases of biphenotypic acute leukemias (BAL), based on the
number and lineage specifity of markers expressed by the blast population (Bene et al.,
Leukemia 9: 1783-6, 1995).
Acute undifferentiated leukemia
An acute undifferentiated leukemia is assumed if only antigens with a low degree of
lineage association such as TdT, HLA-DR, CD7, or CD34 are found in the absence of
lineage specific antigens together with unclassifiable morphology and inconclusive cyto-
chemistry. The ultrastructural detection of MPO may establish the diagnosis of AML-M0 in
a subset of these samples even if MPO is not detected by less sensitive immunological
methods.
Typical antigenic profiles in mature lymphatic neoplasias
As immunophenotyping is currently performed using different methods and antigens no
clear cross correlation of mature lymphoid neoplasias of the B and T lineage to defined
antigenic profiles has been established until now. The revised REAL-classification or
scoring systems, however, have been evaluated, in order to provide immunological pro-
files for the subclassification of mature lymphatic neoplasias in context with their morphol-
ogy. The major goals of immunophenotyping in mature lymphoid neoplasias currently are,
therefore, the assignment of abnormal cells to the B or T cell lineage, their maturational
analysis, and the characterization of specific phenotypes which might be helpful for the
subclassification of disease. In addition, the discrimination of a monoclonal versus reactive
expansion of lymphocytes is a major goal of the analysis.
Leukemic B-cell lymphoproliferative disorders
As a general criterion for the diagnosis of B-lineage non-Hodgkin lymphoma cells the
clonal expression of kappa or lambda light chains should be demonstrated simultaneously
with the expression of pan B cell antigens such as CD19 (Table 8). As an exception plas-
mocytoma cells are characterized by the strong expression of CD38 in the absence of B-
lineage specific antigens or surface immunoglobulins. The cytoplasmic analysis of
immunglobulin light chain expression in CD38-bright cells can help to establish the clo-
nality of plasma cells. A further characteristic immunophenotype is found in hairy cell
leukemia, where B cells with a larger forward scatter signal than normal lymphocytes show
a strong expression of CD11c and CD103. For the diagnosis of chronic lymphocytic
leukemia, CD5 and CD23 in combination with the low intensity of surface immunglobulins
and CD22 are proposed as markers, providing a clear distinction between chronic lym-
phatic leukemia (CLL) and other B-cell diseases.
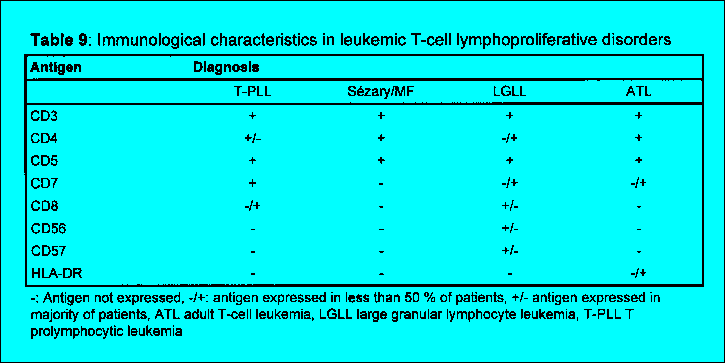
Leukemic T-cell lymphoproliferative disorders
The detection of T-lineage non-Hodgkin lymphoma cells, e. g. in the diagnosis of the
systemic manifestation of a cutaneous T-cell lymphoma is often complicated by a similar
immunophenotype compared to normal T cells (Table 9). Therefore, an unambiguous
immunological diagnosis in T cell lymphoma requires a large excess of cells restricted to a
homogeneous immunophenotype or abnormal expression densities of constitutive anti-
gens when compared to normal T cells.
RECOMMENDED LITERATURE
Argyle JC, Benjamin DR, Lampkin B, Hammond D. Acute nonlymphocytic leukemias in childhood: Inter-observer variability and problems in the use of the FAB classification. Cancer 1989; 63: 295-301.Bene MC, Castoldi G, Knapp W, Ludwig WD, Matutes E, Orfao A, van't Veer MB. Proposals for the immunological classification of acute leukemias Leukemia 1995; 9: 1783-1786.
Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DAG, Gralnick HR, Sultan C. The French- American-British (FAB) Cooperative Group, Proposals for the classification of chronic (mature) B and T lymphoid leukaemias. J Clin Pathol 1989; 42: 567-584.
Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DAG, Gralnick HR, Sultan C. Criteria for the diagnosis of acute leukemia of megakaryocytic lineage (M7). Ann Intern Med 1985; 103: 460-462.
Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DAG, Gralnick HR, Sultan C. Proposed revised criteria for the classification of acute myeloid leukemia. Ann Intern Med 1985; 103: 626-629.
Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DAG, Gralnick HR, Sultan C. Proposal for the recognition of minimally differentiated acute myeloid leukaemia (AML-M0). Br J Haematol 1991; 78: 325-329.
Buccheri V, Matutes E, Dyer MJ, Catovsky D. Lineage commitment in biphenotypic acute leuke- mia. Leukemia 1993; 7: 919-927.
Buccheri V, Mihaljevic B, Matutes E, Dyer MJ, Mason DY, Catovsky D. mb-1: a new marker for B- lineage lymphoblastic leukemia. Blood 1993; 82: 853-857.
Calvelli TA, Denny TN, Paxton H, Gelman R, Kagan J. Guideline for the flow cytometric immuno- phenotyping: a report from the National Institute of Allergy and Infectious Diseases, division of AIDS. Cytometry 1993; 14: 702-714.
Campana D, Coustan-Smith E, Janossy G. The immunologic detection of minimal residual disease in acute leukemia. Blood 1990; 76: 163-171.
Carter PH, Resto-Ruiz S, Washington GC, Ethridge S, Pallini A, Vogt R, Waxdal M, Fleisher T, Noguchi PD, Marti GE. Flow cytometric analysis of whole blood lysis, three anticoagulants, and five cell preparations. Cytometry 1992; 13: 68-74.
Castoldi GL, Liso V, Fenu S, Vegna ML, Mandelli F. Reproducibility of the morphological diag- nostic criteria of acute myeloid leukemia: the GIMEMA group experience. Ann Hematol 1993; 66: 171-174.
Cheson BD, Cassileth PA, Head DR, Schiffer CA, Bennett JM, Bloomfield CD, Brunning R, Gale RP, Grever MR, Keating MJ, et al.. Report of the National Cancer Institute-sponsored Work- shop on Definitions of Diagnosis and Response in Acute Myeloid Leukemia. J Clin Oncol 1990; 8: 813-819.
Creutzig U, Harbott J, Sperling C. Ritter J, Zimmermann M, Löffler H, Riehm H, Schellong G, Ludwig WD. Clinical significance of surface antigen expression in children with acute myeloid leukemia: Results of study AML-BFM-87. Blood 1995; 86; 3097-3108.
Drexler HG, Sperling C, Ludwig WG. Terminal deoxynucleotidyl transferase (TdT) expression in acute myeloid leukemia. Leukemia 1993; 7: 1142-1150.
Drexler HG, Thiel E, Ludwig WD. Review of the incidence and clinical relevance of myeloid anti- gen-positive acute lymphoblastic leukemia. Leukemia 1991; 5: 637-645.
Drexler HG, Thiel E, Ludwig WD. Acute myeloid leukemias expressing lymphoid-associated anti- gens: Diagnostic incidence and prognostic significance. Leukemia 1993; 7: 489-498.
Harris NL, Jaffe ES, Stein H, Banks PM, Chan JKC, Cleary ML, Delsol G, De Wolf Peeters C, Falini B, Gatter KC, Grogan TM, Isaacson PG, Knowles DM, Mason DY, Muller-Hermelink HK, Pileri SA, Piris MA, Ralfkiaer E, Warnke RA. A revised European-American classification of lymphoid neoplasms: A proposal from the International Lymphoma Study Group. Blood 1994; 84: 1361-1392.
Knapp W, Majdic O, Strobl H. Flow cytometric analysis of intracellular myeloperoxidase and lac- toferrin in leukemia diagnosis. In: Ludwig WD, Thiel E (eds.): Recent Advances in Cell Biology of Acute Leukemia: Impact on Clinical Diagnosis and Therapy (Recent Results in Cancer Research, Vol. 131), Springer Verlag, Berlin - Heidelberg, 1993; pp. 31-40.
Lewis DE, Rickman WJ. Methodology and quality control for flow cytometry. In: Rose NR, de Macario EC, Fahey JL, Friedman H, Penn GM (eds.): Manual of Clinical Laboratory Immunol- ogy, 4th ed., chapter 24, American Society for Microbiology, Washington D.C., 1991, pp. 117- 141.
Loken MR, Brosnan JM, Bach BA, Ault KA. Establishing optimal lymphocyte gates for immuno- phenotyping by flow cytometry. Cytometry 1990; 11: 453-459.
Loken MR, Grenier KA, Bach BA. A selected 12-reagent immunophenotyping panel facilitates assignment of lineage in acute leukemia. In: Clinical Monograph No. 3, Becton Dickinson Immunocytometry Systems, 1992; pp. 1-28.
Ludwig WD, Komischke B, Böttcher S. Immunphänotypisierung akuter Leukämien und leukämisch verlaufender niedrig-maligner Non-Hodgkin-Lymphome (Methoden, relevante, Antigene, Interpretation). In: Schmitz G, Rothe G (eds.) Durchflußzytometrie in der klinischen Zelldiagnostik, Schattauer, Stuttgart, 1994; pp. 77-104.
Ludwig WD, Raghavachar A, Thiel E. Immunophenotypic classification of acute lymphoblastic leukaemia. Bailličre's Clinical Haematology 1994; 7: 235-262.
Ludwig WD, Reiter A, Löffler H, Gökbuget, Hoelzer D, Riehm H, Thiel E. Immunophenotypic fea- tures of childhood and adult acute lymphoblastic leukemia (ALL): Experience of the German multicentre trials ALL-BFM and GMALL. Leukemia & Lymphoma 1994; 13 (Suppl. 1): 71-76.
Ludwig WD, Thiel E. Diagnostik der akuten Leukämien mit morphologischen, immunologischen und zytogenetischen Verfahren. Internist 1993; 34: 498-510.
Matutes E, Owusu-Ankomah K, Morilla R, Marco JG, Houlihan A, Que TH, Catovsky D. The immunological profile of B-cell disorders and proposal of a scoring system for the diagnosis of CLL. Leukemia 1994; 8: 1640-1645.
McCoy JP, Carey JL, Krause JR. Quality control in flow cytometry for diagnostic pathology. I. Cell surface phenotyping and general laboratory procedures. Am J Clin Pathol 1990; 93 (Suppl. 1): S27-S37.
National committee for Clinical Laboratory Standards, Clinical applications of flow cytometry: Quality assurance and Immunophenotyping of peripheral blood lymphocytes; Tentative guideline. NCCLS document H42-T (ISBN 1-56238-155-5). NCCLS, Villanova, PA, 1992.
National committee for Clinical Laboratory Standards, Clinical applications of flow cytometry: Immunophenotyping of Leukemic Cells; Proposed Guideline. NCCLS document H43-P (ISBN 1-56238-219-5). NCCLS, Villanova, PA, 1993.
Pinto A, Gattei V, Soligio D, Parravicini C, Del Vecchio L. New molecules at the leukocyte surface: A comprehensive review based on the 5th International Workshop on Leukocyte Differ- entiation Antigens, Boston, USA, 3-7 November, 1993. Leukemia 1994; 8: 347-358.
Pizzolo G, Vincenzi C, Nadali G, Veneri D, Vinante F, Chilosis M, Basso G, Connelly MC, Janossy G. Detection of membrane and intracellular antigens by flow cytometry following ORTHO PermeaFix fixation. Leukemia 1994; 8: 672-676.
Pui CH, Behm FG, Crist WM. Clinical and biologic relevance of immunologic marker studies in childhood acute lymphoblastic leukemia. Blood 1993; 82: 343-362.
Ryan DH. Phenotypic heterogeneity in acute leukemia. Clin Chim Acta 1992; 206: 9-23. Schlossmann SF, Boumsell L, Gilks W, Harlan JM, Kishimoto T, Morimoto C, Ritz J, Shaw S, Silverstein RL, Springer TA, Tedder TF, Todd RF (eds). Leukocyte Typing V: White Cell Dif- ferentiation Antigens. Oxford University Press, London, Oxford, UK, 1995.
Terstappen LWMM, Safford M, Könemann S, Loken MR, Zurlutter K, Büchner Th, Hiddemann W, Wörmann B. Flow cytometric characterization of acute myeloid leukemia. II. Phenotypic het- erogeneity at diagnosis. Leukemia 1992; 6: 70-80.
Terstappen LWMM, Könemann S, Safford M, Loken MR, Zurlutter K, Büchner Th, Hiddemann W, Wörmann B. Flow cytometric characterization of acute myeloid leukemia. I. Light scattering. Leukemia 1991; 5: 315-321.
Terstappen LWMM, Safford M, Unterhalt M, Könemann S, Zurlutter K, Piechotka K, Drescher M, Aul C, Büchner Th, Hiddemann W, Wörmann B. Flow cytometric characterization of acute myeloid leukemia. IV. Comparison to the differention pathway of normal hematopoietic pro- genitor cells. Leukemia 1992; 6: 993-1000.
Traweek ST. Immunophenotypic analysis of acute leukemia. Am J Clin Pathol 1993; 99: 504-512. Urbano-Ispizua A, Matutes E, Villamor N, Sierra J, Pujades A, Reverter JC, Feliu E, Cervantes F, Vives-Corrons JL, Montserrat E, et al.. The value of detecting surface and cytoplasmic anti- gens in acute myeloid leukemia. Br J Haematol 1992; 81: 178-183.
van der Does-van den Berg A, Bartram CR, Basso G, Benoit YCM, Biondi A, Debatin KM, Haas OA, Harbott J, Kamps WA, Köller U, Lampert F, Ludwig WD, Niemeyer CM, van Wering ER. Minimal requirements for the diagnosis, classification, and evaluation of the treatment of childhood acute lymphblastic leukemia (ALL) in the "BFM Family" cooperative group. Med & Pediat Oncol 1992; 20: 497-505.
van´t Veer MB. The diagnosis of acute leukemia with undifferentiated or minimally differentiated blasts. Ann Hematol 1992; 64: 161-165.
van´t Veer MB, Kliun-Nelemans JC, van der Schoot E, van Putten WLJ, Adriaansen HJ, Wering ER. Quality assessment of immunological marker analysis in the diagnosis of leukemia nad lymphoma: a multicenter study. Br J Haematol 1992; 10: 450-465.
van Dongen JJM, Breit TM, Adriaansen HJ, Beishuizen A, Hooijkas H. Detection of minimal resid- ual disease in acute leukemia by immunological marker analysis and polymerase chain reac- tion. Leukemia 1992; 6: 47-59.
Verschuren MC, Comans-Bitter WM, Kapteijn CA, Mason DY, Brouns GS, Borst J, Drexler HG, van Dongen JJM. Transcription and protein expression of mb-1 and B29 genes in human hematopoeitic malignancies and cell lines. Leukemia 1993; 7: 1939-1947.
Verwer BJ, Terstappen LWMM. Automatic lineage assignment of acute leukemias by flow cyto- metry. Cytometry 1993; 14: 862-875.
Vidriales MB, Orfao A, López-Berges MC, González M, López-Macedo A, Garcia MA, Galende J, San Miguel JF. Light scatter cahracteristics of blast cells in acute myeloid leukaemia: association with morphology and immunophenotype. J Clin Pathol 1995; 48: 456-462.
Wörmann B, Grove D, Terstappen LWMM. Multiparametrische Charakterisierung von Leukämie- und Lymphomzellen. In: Schmitz G, Rothe G (eds.) Durchflußzytometrie in der klinischen Zelldiagnostik, Schattauer, Stuttgart, 1994; pp. 105-115.
Wörmann B, Safford M, Könemann S, Büchner Th, Hiddemann W, Terstappen LWMM. Detection of aberrant antigen expression in acute myeloid leukemia by multiparameter flow cytometry. Recent Results Cancer Res 1993; 131: 185-196.
ABBREVIATIONS
ACD-A acid citrate dextrose, solution AALL acute lymphoblastic leukemia
AML acute myeloid leukemia
AML-M0 myeloblastic leukemia without maturation
AML-M1 myeloblastic leukemia with minimal maturation
AML-M2 myeloblastic leukemia with maturation
AML-M3 hypergranular promyelocytic leukemia
AML-M4 myelomonocytic leukemia
AML-M5 monocytic leukemia: (M5a) poorly differentiated, (M5b) well differentiated
AML-M6 erythroleukemia
AML-M7 megakaryoblastic leukemia
ATL adult T-cell leukemia
AUL acute undifferentiated leukemia
BSA bovine serum albumin
CC centrocytic Non-Hodgkin lymphoma
CD cluster of differentiation as defined by the 5th CD workshop
CLL chronic lymphatic leukemia
cyCD3 cytoplasmic expression of CD3
cyCD22 cytoplasmic expression of CD22
cyCD68 cytoplasmic expression of CD68
cyIgµ cytoplasmic expression of immunoglobulin µ-chain
EDTA ethylenediaminetetraacetic acid
F1 fluorescence 1
F2 fluorescence 2
F3 fluorescence 3
FAB French-American-British cooperative group (see Bennet et al., 1985-1991)
FCS fetal calf serum
FITC fluorescein isothiocyanate
FL follicular lymphoma
FS forward scatter
GpA glycophorin A
HCL hairy cell leukemia
LGLL large granular lymphocyte leukemia
LP-IC lymphoplasmocytic immunocytoma
mAb monoclonal antibody
mAbs monoclonal antibodies
MCL mantle cell lymphoma, centrocytic lymphoma
MDS myelodysplastic syndrome
MPO myeloperoxidase
NHL Non-Hodgkin's lymphoma
NK cells natural killer cells
PBS phosphate buffered saline
PCL plasma cell leukemia
PLL prolymphocytic leukemia
PMT photomultiplier
R-PE R-phycoerythrin
R-PE/Cy5 R-phycoerythrin and Cy5 tandem conjugate
Sézary/MF Sézary syndrome / mycosis fungoides
SIg surface membrane immunoglobulins
SS side scatter
TcR T-cell receptor
TdT terminal deoxinucleotidyl transferase
T-PLL T prolymphocytic leukemia
APPENDIX
Methods for sample preparation(1) Direct incubation with mAbs followed by erythrocyte lysis
In this method the material in a first step is directly incubated with the fluorochrome- conjugated mAbs. Typically 0.3 to 1 x 106 nucleated cells are incubated in a total volume 100 of 200 µl (bone marrow or blood supplemented with staining buffer dependent on cell counts) simultaneously with all fluorochrome-conjugated mAbs for 15 to 30 min either at 4 C or at room temperature in the dark. Incubation at room temperature may allow a shorter incubation time. However, at room temperature azide has to be supplemented in order to inhibit capping or internalization of the mAbs.
In a second step, erythrocytes are lysed using ammonium chloride solutions, hypotonic sodium chloride solutions, or commercial lysing solutions according to published methods or manufactures description. After removal of unbound mAbs by washing, the cell sus- pension can be either fixed or directly analyzed by flow cytometry.
If surface immunoglobulin expression or other antigens also present in high quantities in plasma are analyzed using this method, the whole blood or bone marrow has to be washed twice in the staining buffer before incubation with the mAbs in order to remove the excess of immunoglobulins in plasma.
(2) Erythrocyte lysis followed by incubation with mAbs
In this method in a first step erythrocytes are lysed by incubation with ammonium chlo-
ride solutions or hypotonic sodium chloride solutions. Most commercial lysing solutions
contain a low concentration of fixants. Fixation can lead to the destruction of epitopes and
should be avoided before incubation with mAbs. After erythrocyte lysis the cells are
washed twice, counted, and incubated at 0.3 to 1 x 106 cells in 100 µl of staining buffer
simultaneously with all fluorochrome-conjugated mAbs for 15 to 30 min either at 4 C or at
room temperature in the dark. Incubation at room temperature may allow a shorter incu-
bation time. However, azide has to be used at room temperature to inhibit capping or
internalization of the mAbs. After removal of unbound mAbs by washing the cell suspen-
sion can be either fixed or directly analyzed by flow cytometry.
This procedure is associated with a higher number of washing steps when compared to the alternative lysis procedure (1). As an advantage, however, a constant number of nu- cleated cells can be resuspended in a constant volume, e.g. 0.3 to 1 x 106 cells per 100 µl PBS for immunostaining even if the initial concentration of nucleated cells is low in the material analysed. Furthermore, the analysis of surface immunoglobulin expression is feasible without separate preparation steps.
(3) Density gradient preparation of mononuclear cells
In this method in a first step mononuclear cells are isolated as the interphase following
centrifugation of diluted bone marrow or blood on top of a separation medium (d=1,077).
The cells are washed followed by a facultative red cell lysis step (red cell contamination
may occur upon preparation of old specimens) and incubation of the washed cell fraction
at 0.3 to 1 x 106 cells in 100 µl of staining buffer simultaneously with all fluorochrome
conjugated mAbs for 15 to 30 min either at 4 C or at room temperature in the dark. Incu-
bation at room temperature may allow a shorter incubation time. However, azide has to be
used at room temperature to inhibit capping or internalization of the mAbs. After removal
of unbound mAbs by washing the cell suspension can be either fixed or directly analyzed
by flow cytometry.
If the analysis can not be performed immediately, the isolated mononuclear cell sus- pension may be stored in a tissue culture medium at 4 C. It is suggested to prepare a smear following preparation of the mononuclear cell fraction in order to control the com- position of the isolated cell fraction.


Fig.1 Scheme of the immunophenotype of myeloid cells during differentiation. The grey bars mark mature cellular phenotypes physiologically found in the peripheral blood compartment.
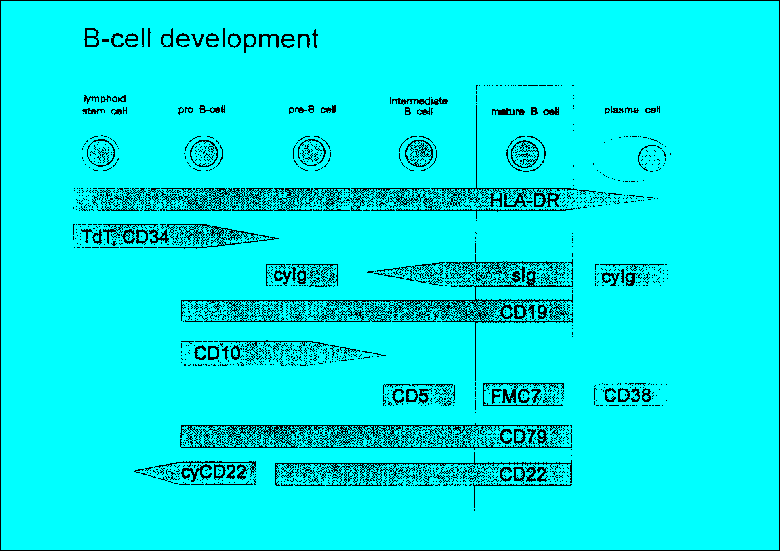

Fig.2 Scheme of the immunophenotype of lymphoid cells during differentiation. The grey bars mark cellular phenotypes physiologically found in the peripheral blood compartment.
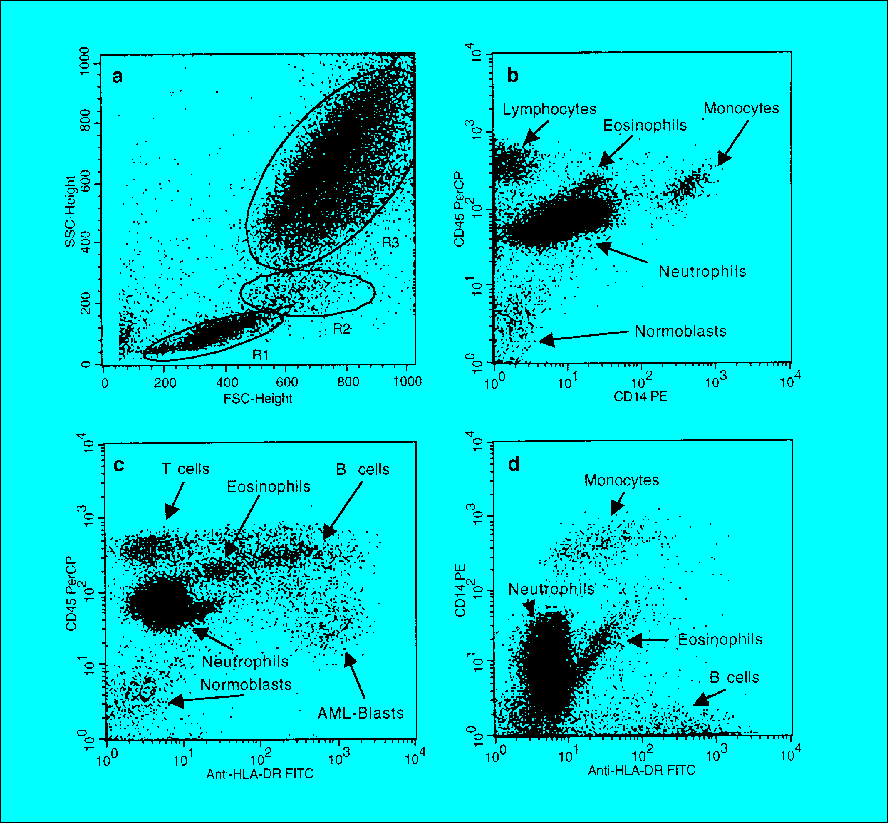
Fig.3 Definition of analysis regions based on the light scatter characteristics of normal mature peripheral blood leukocytes based on the combination of cellular forward and side scatter characteristics (a) with surface expression of CD45 and CD14 (b). Region 1 defined based on CD45-bright and CD14-negative lymphocytes will in addition contain immature blast cells, normoblasts and basophils, region 2 defined based on CD14-bright monocytes will in addition contain immature blast cells, normoblasts and basophils, and region 3 defined based on CD45-dim neutrophils will contain more mature myeloid elements. The simultaneous analysis of HLA-DR (a and d) is helpful in the identification of normal and abnormal precursor cells within regions 1 and 2
Tab.1 Antigens frequently analyzed in the immunophenotyping of hematopoietic malignancies

Tab.2 Characterization of antigens
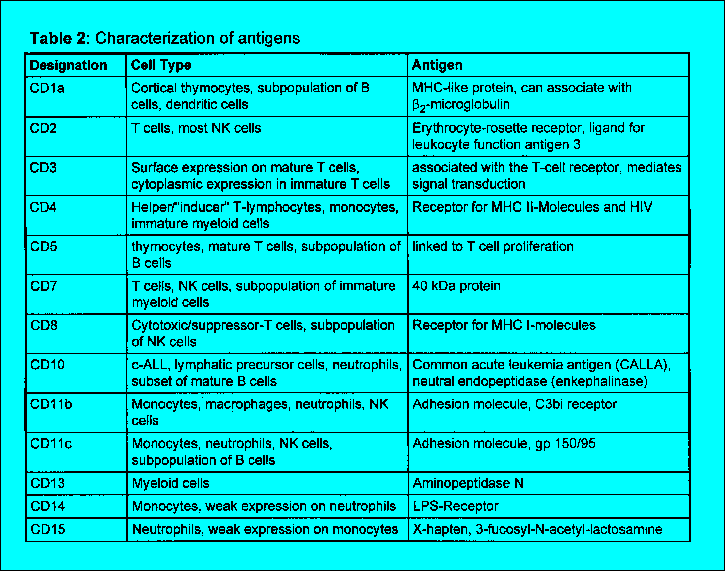
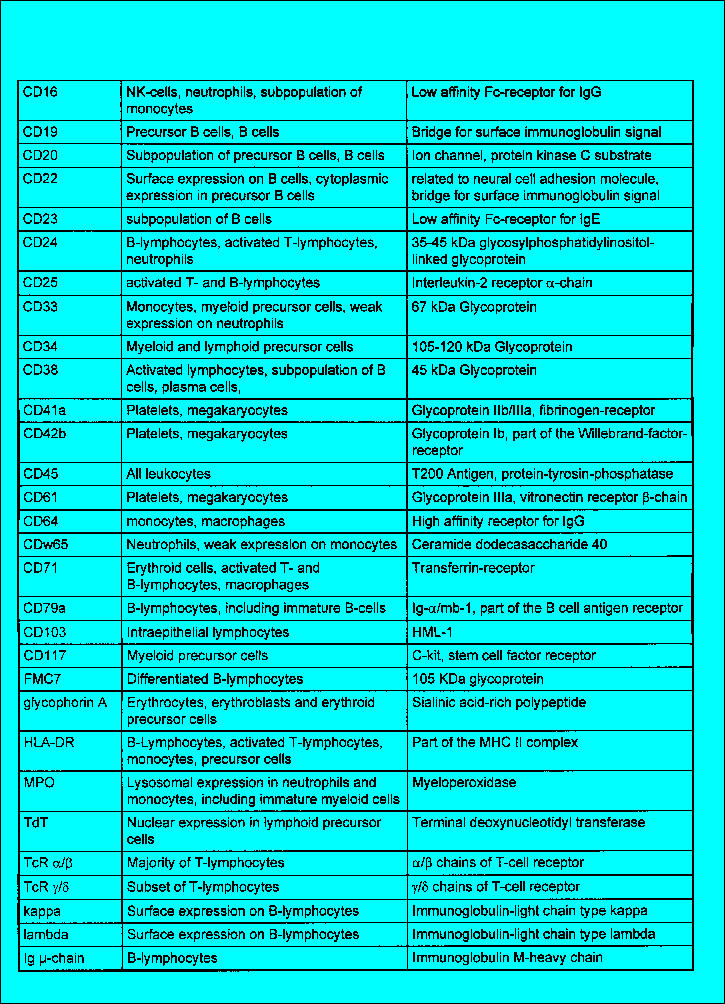
Tab.3 Multiparametric phenotypes of normal blood and bone marrow cells
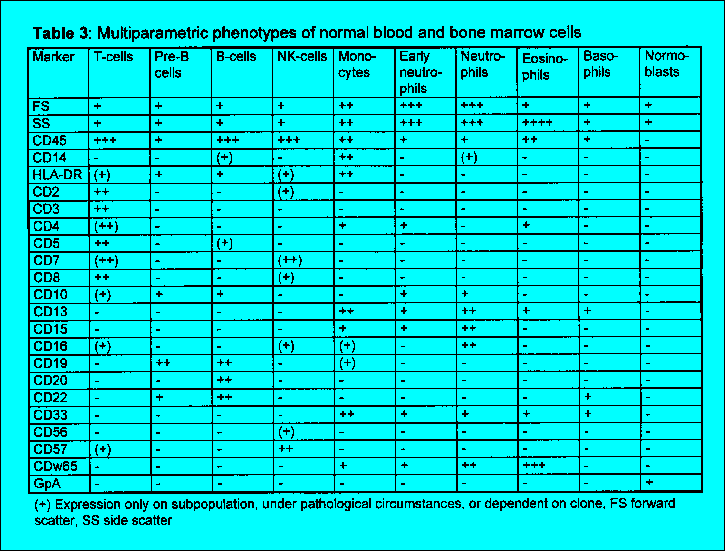
Tab.4 Consensus on two-colour immunophenotyping in acute leukemia
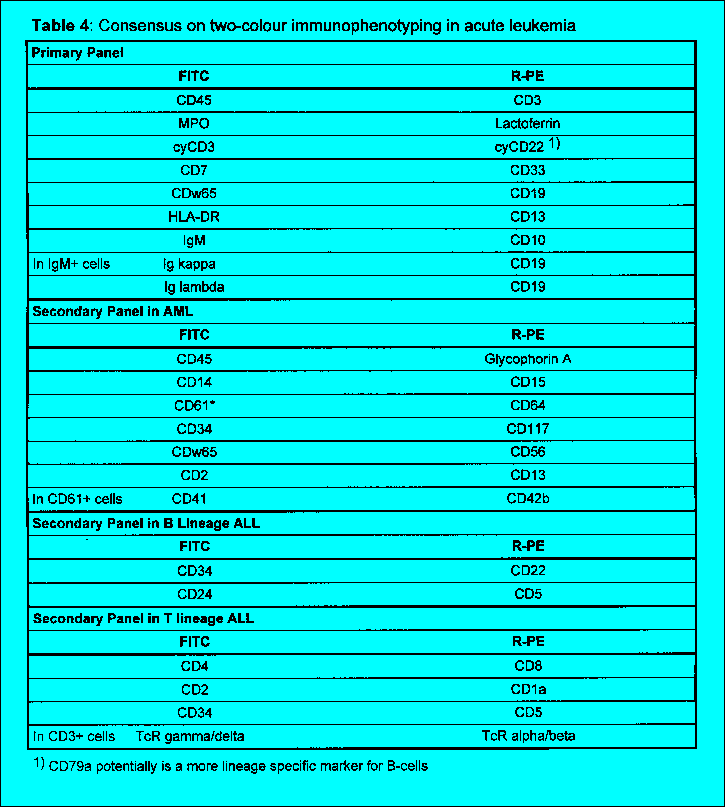
Tab.5 Consensus on two-colour immunophenotyping in mature lymphoid neoplasias
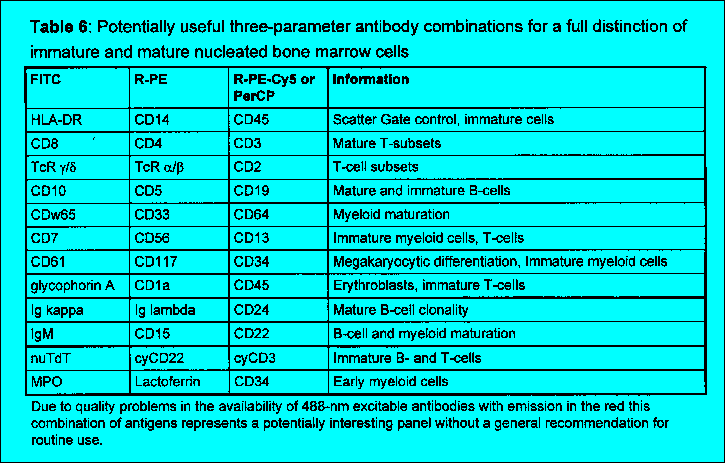
Tab.6 Potentially useful three-parameter amtibody combinations for a full distinction of immature and mature nucleated bone marrow cells
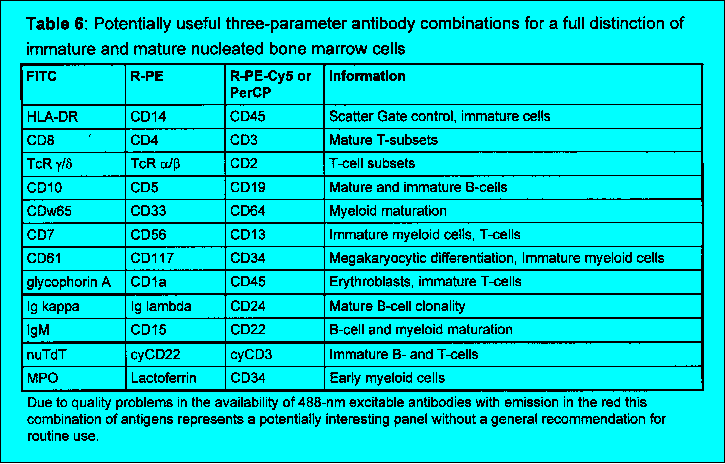
Tab.7 Immunological characteristics in acute myeloid leukemia
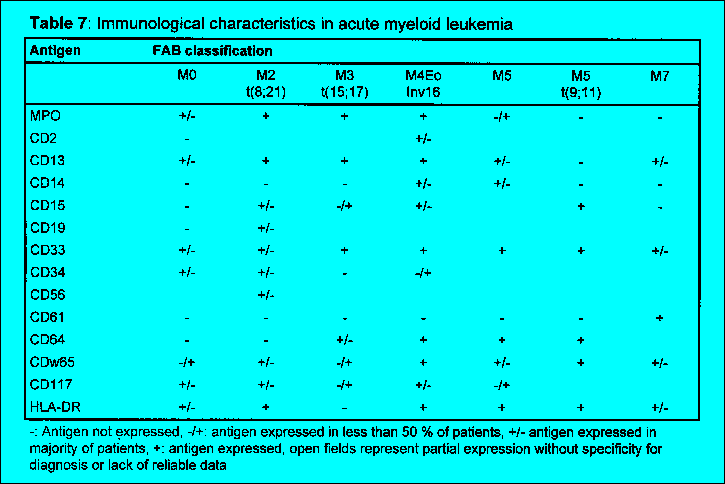
Tab.8 Immunological characteristics in leukemic B cell
lympho-proliferative disorders

Tab.9 Immunological characteristics in leukemic T cell
lympho-proliferative disorders