

Flow cytometry is concerned with the measurement of the light intensity of a cell whether it be scattered laser light or fluorescence emitted by a fluorochrome. Light is detected by a photomultiplier tube (PMT) or a photodiode which converts it via a pre-amplifier to a voltage ie an electrical output that is proportional to the original fluorescence intensity. Depending on the range of the signals, the voltage is then input into either a linear or logarithmic amplifier.
The use of a logarithmic amplifier is indicated in most biological situations where distributions are skewed to the right. In this case the effect of the log amp is to normalise the distribution - it is said to be Log Normal and the data has been log-transformed. The use of a log amp is also required when there is a broad range of fluorescence as this can then be compressed; again this is true of most biological distributions. Linear amplification is used when there is not such a broad range of signals e.g. in DNA analysis and calcium flux studies. The shape of a logarithmically transformed distribution will be unchanged no matter where it is located along the scale, wheras a linearly amplified distribution will be broader towards the right of the x axis.
So it is the setting of the amplifier as either logarithmic or linear that will determine how the signals from the cells are amplified. These output voltages, which are a continuous distribution, are converted to a discrete distribution by an Analog to Digital converter (ADC) which places each signal into a specific channel depending on the level of fluorescence. Once the data have been acquired, the shape of a particular distribution is fixed.
To convert a continuous data distribution that is acquired from cells into a discrete distribution the ADC forms a histogram where the x axis is divided into a certain number of channels. This is either 256 or 1024 channels depending on the type of ADC used (an 8-bit ADC gives 28 i.e. 256 channels, a 10-bit ADC gives 210 i.e. 1024 channels - 10 bit ADCs are more common and will be considered in the following examples). So each data point will fall into a particular channel depending on its level of fluorescence. To get that value we need to look at the scale of the x axis. Regardless of whether data have been acquired using a linear or logarithmic amplifier, histograms can be displayed on a linear or a logarithmic scale. The scale itself does not affect the raw data - the histogram depends on the amplifier that collected the data - but will affect any derived data e.g. mode, mean, median etc.
If the scale is linear, the display is as channel numbers i.e. 0-1023; if it is logarithmic (a 4 decade log amp), it is as linear values ie 1-10,000 (just to make things crystal clear!). For data that have been acquired by linear amplification, channel numbers and linear values are equivalent, but for log amplified data we can choose either channel numbers or linear values. We can relate the two: the log amps used in the flow cytometer are generally 4 decade logs so the range is 100-104 and each log decade takes up a quarter of the available channels.
Therefore:
Decade Channels Linear values
100-101 0-255 1-10
101-102 256-511 10-100
102-103 512-767 100-1000
103-104 768-1023 1000-10000
The linear value for a particular channel number can be calculated by using:
10 channel no/256
or vice versa, a linear value can be converted to a channel number by:
log(linear value) x 256
The rule of thumb to use is: linear amplified data (DNA is the most important application) should be displayed as channel numbers; logarithmically amplified data (the majority of immunofluorescence work) can be either channel numbers or linear values. The decision as to which to use will depend on what you want to get from the experiment but the most common option will be to use linear values (i.e. a log scale). Let us look at a couple of examples.
The simplest type of experiment involves using an immunofluorescent marker to look for a positive sub-population of cells. In this case the percentage of cells expressing the marker can easily be determined by using a marker (below) and if the percentage of positive cells is all that is required the x-axis scale is immaterial.
However, things become more complicated when we want to get some idea of the level of fluorescence. In the example below the two distributions show different levels of fluorescence.
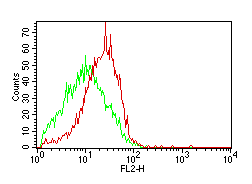
How can these differences be quantified?
To quantify flow cytometric data we need to look at the measures of the distribution of a population. The measures of central tendency are the mode, the mean and the median. How can each of these be used?
The mode is the channel with the most events in. However, it is rarely used as it is subject to data blips especially if there is a build-up of data in the first or last channel.
The mean is the "average" and can be either arithmetic or geometric. The arithmetic mean is calculated as Sigma(x)/n, and the geometric mean as n root(a1 x a2 x a3....an). In general, with log-amplified data the geometric mean should be used as it takes into account the weighting of the data distribution, and the arithmetic mean should be used for linear data or data displayed on a linear scale.
The median is the central value i.e. the 50th percentile, where half the values are above and half below.
The mean and median can both be used as measures to quantitate cellular fluorescence. In a linearly amplified distribution there are rarely problems as the mean and median are easily calculated, but this is not the case with log amplified data and problems can arise here. As we have seen we can use either linear values or channel numbers; how can we use the mean and median to relate levels of fluorescence intensity?
To compare absolute fluorescence values, it is best to use linear values as these can be directly compared i.e. a cell with a linear value of 100 is 10 times brighter than a cell with a linear value of 10. This cannot be done with channel number - a cell in channel 512 is still 10 times as bright as one in channel 256- but we should talk about "channel shifts" i.e. a shift in fluorescence intensity of 256 channels.
It is important to decide before you start analysing, what you want out of the experiment. The choice of amplification and method of analysis should then be logically determined.
Logic and consistency are the keys to successful data analysis in flow cytometry, and realising that at best flow cytometry is only semi-quantitative. It is excellent for relative comparisons but more problematic when asking for absolute quantitative data.