
Single colour DNA Staining
The most commonly used DNA dye is propidium iodide (PI), which intercalates in the DNA helix and fluoresces strongly red (emission maximum 637nm). It has the advantage that it is excited by 488nm light and can be used on most common benchtop flow cytometers. However it does require cells to be fixed or permeabilised and therefore non-viable. PI also stains double-stranded RNA and this should be removed with ribonuclease.
An alternative is to employ Hoechst 33342 which binds to AT-rich regions in the DNA and will enter viable cells without the need for fixation, so cells can be recovered and grown afterwards. The rate of dye uptake is dependent on dye concentration and cell type. The disadvantage with this method is that Hoechst 33342 is UV-excited so cannot be used on most benchtop flow cytometers. However it may allow dye combinations that are not possible with propidium iodide.
These two dyes are the standards used here at ICRF.
The protocol for DNA staining by propidium iodide is as follows:
![]() After harvesting and washing cells, fix in ice-cold 70% ethanol (ethanol in distilled water) while vortexing. Samples can stay in ethanol for up to 7 days at 4°C.
After harvesting and washing cells, fix in ice-cold 70% ethanol (ethanol in distilled water) while vortexing. Samples can stay in ethanol for up to 7 days at 4°C.
![]() Spin at 2000rpm, 5 mins. Wash x2 in PBS.
Spin at 2000rpm, 5 mins. Wash x2 in PBS.
![]() Treat cells with 100Ál of100Ág/ml ribonuclease for 5 minutes at room temperature.
Treat cells with 100Ál of100Ág/ml ribonuclease for 5 minutes at room temperature.
![]() Add 400Ál propidium iodide (50Ág/ml). Analyse by flow cytometry using 488nm excitation, gating out doublets and clumps using pulse processing and collecting fluorescence above 620nm. A typical profile looks like this.
Add 400Ál propidium iodide (50Ág/ml). Analyse by flow cytometry using 488nm excitation, gating out doublets and clumps using pulse processing and collecting fluorescence above 620nm. A typical profile looks like this.
The protocol for DNA staining by Hoechst 33342 is as follows:
![]() Treat the cells with Hoechst 33342 for 10-60 minutes at 37°C. The concentration of Hoechst used must be pre-determined, as the optimal concentration will vary with cell type, but will probably be in the range 5-20Ág/ml.
Treat the cells with Hoechst 33342 for 10-60 minutes at 37°C. The concentration of Hoechst used must be pre-determined, as the optimal concentration will vary with cell type, but will probably be in the range 5-20Ág/ml.
![]() Spin at 1000rpm, 5 mins. Wash x2 in PBS. If appropriate add propidium iodide to identify dead cells.
Spin at 1000rpm, 5 mins. Wash x2 in PBS. If appropriate add propidium iodide to identify dead cells.
![]() Analyse by flow cytometry, using ultra-violet and 488nm excitation, collect emitted Hoechst fluorescence between 390nm and 480nm.
Analyse by flow cytometry, using ultra-violet and 488nm excitation, collect emitted Hoechst fluorescence between 390nm and 480nm.
PULSE PROCESSING
Most DNA profiles can be improved if cell doublets are identified and excluded from analysis. This is done by analysing the pulse produced as a particle passes through the laser beam. Two G1 cells stuck together will appear to have the same amount of DNA as a single G2 cell. However, since they are bigger they will take slightly longer to pass through the laser beam, so by analysing pulse width versus pulse height or area we can eliminate the majority of doublets from the anlaysis. This example shows the effect of removing cell doublets from the analysis
Permeabilisation with, for example, saponin, will also enable internal antigen staining and will give acceptable DNA profiles. Therefore some preliminary experiments may be required to determine the best method. We have found that many antigens may be stained with FITC-labelled antibodies and then fixed in 70% ethanol. Analysis then enables the percentage of cells expressing the antigen to be determined and the DNA profiles of positive and negative cells to be analysed.
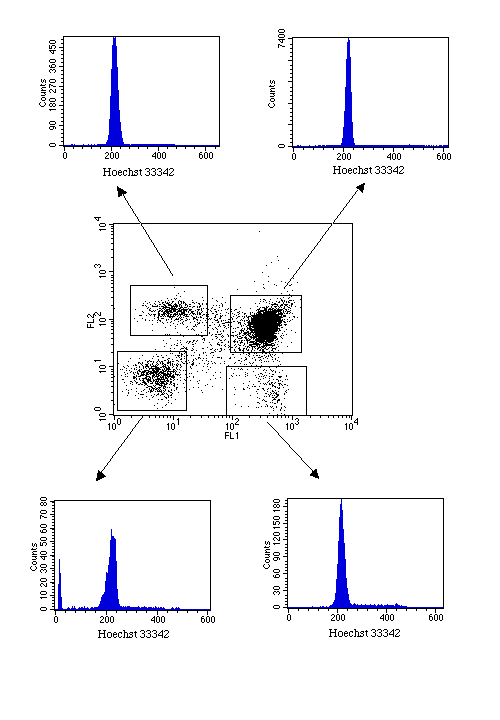
This example is of CD2-transfected cells.
DNA plus antigen staining is also possible using Hoechst 33342, although this does require a dual laser set-up. However, it does have the added advantage that, depending on the fluorochromes used for antigen detection, dead cells may also be excluded from the analysis. Using this method, the cell cycle status of phenotypically-defined sub-populations can be assessed.
{kind=link}
{kind=link}
{kind=link}
{kind=link}