Introduction
This chapter is dedicated to the principal techniques to analyze the cytoskeleton, the most appropriate being the immunocytochemical methods which will be herein detailed. Other techniques, such as DNA transfection and microinjection, which also offer a valid contribution to the analysis of the cytoskeleton in fluorescence microscopy, will be only briefly introduced in this chapter. For further information, we suggest to consider ref. 2 (for DNA transfection) and ref. 3 (for microinjection).
DNA transfection follows the cloning of a gene, reintroducing it into various eukaryotic cell lines and thus allowing the study of the protein-encoded characteristics. In fact, by using different techniques, cells can be induced to internalize the gene of interest expressed by eukaryotic vector. Both normal or manipulated genes can be transfected in cells and, by this way, transfection represent a very useful method to elucidate structure-function relationships. Microinjection allows access of various molecules to cells like the ones to which cells are impermeable, antibodies for intracellular proteins and cellular proteins fused to fluorescent markers. Whatever the way used to introduce such molecules (glass micropipette, vesicle etc.), this technique offers the possibility of studying the function of a protein as well as the dynamic of some structures.
However, as stated above, the elective experimental methods for studying the cytoskeleton during apoptosis are immunocytochemical techniques (4), such as fluorescence microscopy, flow cytometry and immunoprecipitation (5). They will offer useful approaches to study qualitative and quantitative alterations of the cell cytoskeleton as well as of a number of proteins regulating the cytoskeleton state, also in response to an apoptotic stimulus.
This chapter can be useful considering that the field we are dealing
with has developed so rapidly in the last few years that a novice may encounter
difficulty selecting an appropriate technique, performing the various steps
of the procedure correctly, and interpreting results. The following three
techniques will be detailed:
Fluorescence microscopy
A.1 Introduction
A.2.2 Methods
Standard protocol
Perform steps from 1 to 3 only for floating cells. For cells grown on coverslips consider step 4 as the first.
Protocol advised for polymerized actin (F-actin) detection
A3. Commentary
A3.1. Background Information
2. The choice of an appropriate experimental protocol for detecting
the cytoskeletal proteins of interest.
Other problems that we can encounter by using this technique are the following:
False-negative results
- Modification or loss of the antigenic sites during fixation. In this case can be useful to modify the fixative solution or the kind of fixative used.
- Lack of antibody penetration for incomplete permeabilization. It is possible bypass this problem prolonging the time of step 7 of the standard protocol or to use a more concentrated solution of permeabilizing agent.
- Sterical hindrance or the masking of antigenic sites by proteins. In this particular case it can be useful to use, after cell fixation, a detergent solution for a few minutes. Wash and then go on as indicated in the standard protocol.
- Presence of free reactive groups after aldehyde fixation. These groups can be still present into the cells leading to non-immunological binding of proteins (e.g. IgGs). This can be prevented by incubation of the cells with serum not recognized by the immunoreagents or by the use of buffers containing small molecules (lysine, glycine, tris) or proteins (gelatine, serum albumin), both with reactive amino groups.
- Hydrophobic and/or ionic interactions. Ionic interactions of antibodies (Fc region) with cellular components such as nuclear and ribosomal components, can be minimized by the use of buffer with enhanced ionic strength. Hydrophobic interactions can result from hydrophobic parts present in antibodies and labels with hydrophobic cellular compounds. The addition of detergents in washings fluids and incubation media might reduce this interaction. Moreover, to minimize the influence of ionic charges, it was found that incubating the tissue section at pH 8.6 results in a decrease in background levels. The rationale behind this treatment is that IgGs have an isoelectric point of 8.6, which means that incubation at this pH results in a minimal charge on the IgG molecules.
- Autofluorescence of cellular components such as flavonoids etc.
- Although nonspecific staining may be avoid with precautionary measures, each study dealing with a particular antigen should include a series of controls in order to ensure that non specific localization occurred. The most obvious control is one that uses normal serum in place of, and from the same animal source as, the primary antibody. While this does not preclude the possibility that the primary antibody is staining something other than the antigen being sought, it does give some indication of back-ground or nonspecific staining due to other components in the tissue or components in the reagents being used. In addition to normal serum, both negative and positive control tissue should be used in order to detect discrepancies from one experiment to the next. The use of antibodies directed against irrelevant antigens is also helpful in determining whether the animal used for antibody production contains naturally occurring, or nonspecific, antibodies against irrelevant antigens other than the one under study. In such cases, localization would be expected to occur regardless of primary antibody specificity. The absorption of primary antibody along with its antigen is perhaps the most definitive control, but this occurrence is not an absolute guarantee that the correct antigen has been localized. This lack of certainty can best be explained by examining the binding of monoclonal antibodies. Such antibodies are against a very small portion of the protein, amounting to approximately seven amino acids. It is not unreasonable to imagine that more than one protein in a given tissue may contain a similar sequence of seven amino-acids. Still, the use of appropriately purified reagents and sufficient controls can greatly reduce the opportunity for error.
Phosphate Buffer Solution (PBS) 1x
For preparation of coated coverslips: deposit 40 m l of this solution on 13 mm coverslips and wait the complete evaporation. Store the coverslips at room temperature protecting them from the dust.
Appendix A2
The quantification of cytoskeletal proteins can be obtained by using flow cytometry. Measurements of fluorescent dyes bound to cellular constituents represent the majority of flow cytometric applications. Fluorescence has three major advantages in flow studies: i) fluorescence emission is direcltly proportional to specific cell constituents; ii) low concentration of dye per cell can be detected; and iii) nonfluorescent compounds can be made fluorescent by intracellular enzymes.
B2. Protocol
B2.1. Materials
Standard protocol for cytoskeletal proteins' detection by flow cytometry
Repeat steps from 1 to 4 of the standard protocol.
6. Add 200 m l of Triton solution (see Appendix B1) for 10 min at 4°C.
7. Wash cells twice with cold PBS buffer.
8. Centrifuge cells at 800 rpm for 5 min.
9. Resuspend cells in 1 ml of 1% AB human serum in cold PBS.
10. Wash cells with PBS buffer.
11. Resuspend cells in 50 m l of 1% AB human serum in cold (see Appendix B1).
12. Incubate cells with specific antibody for 1 hour at 4°C.
13. Wash cells twice with PBS buffer.
14. For the indirect method, incubate the cells with labelled second antibody for 30 min at 4°C. For the direct method go directly to step 16.
15. Wash cells twice with PBS.
16. Resuspend cells in 500 m l of cold PBS.
17. Analyze with a cytofluorimeter.
B3.1. Background Information
See A3.1
B3.2. Critical Parameters
See A3.2.
B3.3. Troubleshooting
The quantitative alterations of cytoskeletal components, such as for example polimerized actin, can in certain cases be associated with cell death hinderance (10). It is fundamental, however, to distinguish between real changes in quantity of the cytoskeletal proteins and artefacts due to inappropriate experimental procedures. For this reason is very important to adjust the cytometer by using the specific negative controls.
All problems relative to flow cytometry tecnique are discussed in detail in the chapters of this book specific for flow cytometry.
Phosphate Buffer Solution (PBS) 1x
KCl 0.25 g
NaHPO4 x 2 H2O 1.44 g
KH2PO4 0.25 g
Add 1 L of distilled H2O.
pH from 7.2 to 7.4.
Store at 4°C for a few weeks.
in PBS pH 7.4.
Store at 4°C for a few months.
in PBS pH 7.4
Store at 4°C for a few months.
in PBS pH 7.4.
Store at 4°C in the dark for a few days.
in PBS pH 7.4.
Store at room temperature for a few weeks.
in PBS pH 7.4.
Store at 4°C for few days.
Appendix B2
3. Methyl alcohol Carlo Erba (cat. n. 414816)
4. Nonidet P-40 Sigma (cat. n. N-6507)
5. Paraformaldehyde ICN (cat. n. 17538)
6. Triton X-100 Sigma (cat. n. 9002-93-1)
7. Trypsin ICN (cat. n. 101179)
2. Cytometer, equipped with a computer.
Recent advances in such a field combined DNA transfection with immunoprecipitation, thus offering a contribution to the analysis of proteins in general and of proteins of the cytoskeleton in particular (see for example ref 7). A clear improvement of the coupling of these two techniques is the fact that not all proteins have antibodies capable of immunoprecipitating, thus it is possible to manipulate the gene introduced by transfection so that it can bring an artificial epitope. For example, one of the most useful epitope utilized is the protein G of VSV, recognized by the monoclonal antibody P5D4. By introducing a protein tagged by this epitope, the protein itself acquire the capacity of being immunoprecipitated, thus rendering easier the study of the protein characteristics.
- lysis of the cells to release the antigen and preclearing of lysates
- formation of the antibody-antigen complex
- purification of the immune complex
2. NaCl
3. Trizma base
4. Protein A/G Agarose
5. Leammli buffer
Lysis of cells and preclearing lysate
2. Incubate cells in lysis buffer A (107 cells in 1 ml buffer prechilled to 4°C) for 1 h at 4°C.
3. Scrape the cells and debris from the plate with a rubber policeman and transfer all the preparation to a 1.5 ml conical tube.
4. Centrifuge 10 min at 10,000 g at 4°C.
5. (Optional) Centrifuge the supernatant at 100,000 g for 30 min.
6. Quantify the protein concentration and use the same amount of proteins for all samples.
7. Preclear supernatant by adding 20 m l of control Protein A/G agarose prepared without antibody or coupled to non specific antibody.
8. Shake on an orbital shaker for 1 h at 4°C.
9. Centrifuge 1 min at 10,000 g and save the supernatant.
(Optional from 7 to 9)
11. Incubate 1 h on ice
13. Centrifuge at 10,000 g for 1 min at 4°C.
14. Remove the supernatant by aspiration and add 0.5 ml of lysis buffer. Vortex to resuspend.
15. Wash three times with lysis buffer. Remove the last wash as completely as possible.
16. Add 20 m l of Leammli sample buffer 1X. Heat for 5 min at 100°C, centrifuge at full speed and use the released immune complex for SDS-polyacrilamide gel electrophoresis (SDS-PAGE) (5).
17. Detection of the immunoprecipitation reaction can be performed with the following methods:
a) Comassie Brilliant Blue or Silver staining (ref 5) if the amount of protein is >1 m g.
b) Transfer to nitrocellulose and blotting (ref 5) if the amount of proteins is <1 m g.
c) Autoradiography (ref 6) if you labelled with radioactive amino acid to detect very rare proteins
C3.1. Background information
In itself, this technique is a very simple one, but it takes time and accuracy to find the right working conditions, almost every antigen requires a particular protocol "treatment".
2. In general, the conditions used for lysis should be as gentle as
possible to retain the antibody binding site and to avoid solubilizing
background proteins, but they have to ensure quantitative release of the
antigen. Salt concentration, type of detergent, presence of divalent cations
and pH are all variables that affect the release of polypeptide antigens.
The following parameters should be monitored to determine the optimal conditions
of extraction:
- non ionic detergent concentrations between 0.1 and 2%
- ionic detergent concentrations between 0.01 and 0.5%
- divalent cations concentrations between 0 and 10 mM
- pH between 6 and 9
3. A gel run after immunoprecipitation contains not only the protein of interest, but also the antibodies as one of the major band. Antibodies are formed by light and heavy chain which together have a molecular weight of about 150 kDa. The light chain alone is at the MW of many important proteins which are usually studied by immunoprecipitation. Thus, in the case of Comassie blue, silver staining and immunoblotting is recommended to use a Laemmli buffer without DTT so that the two chains are not separated during migration and they can be found as a unique band at almost 150 kDa. Also, to avoid this problem in immunoblotting, different antibodies might be used to immunoprecipitate and to detect the protein after transfer, i.e. polyclonal antibodies to immunoprecipitate and monoclonal antibodies to detect the protein. Thus, the secondary antibody will be an anti-mouse which will not recognize the polyclonal immunoglobulin used to immunoprecipitate.
The most frequent problem is the presence of non-specific background which may be reduced by the following recommendations:
1. Perform the preclearing step, reported in the text as optional.
2. Add proteins such as BSA, gelatine to the lysate
3. Preincubate protein A/G Agarose with 2% BSA prior adding to lysate
4. Use more stringent washes such as 1 M NaCl.
5. Increase the number of wash cycles
6. Try a different antibody
Various works have been published so far in which both types of proteins have been immunoprecipitated. In particular see ref. 8.
Phosphate Buffer Solution (PBS) 1x
KCl 0.25 g
NaHPO4 x 2 H2O 1.44 g
KH2PO4 0.25 g
Add 1 L of distilled H2O.
pH from 7.2 to 7.4.
Store at 4°C for a few weeks.
Na Cl 150 mM
Triton X-100 0.5% (see also critical parameters)
Store at 4°C for a few weeks
Trizma base 60 mM
glycerol 10%
DTT 100 mM
bromophenol bleu 0.1%
Store at -20°C for months
Methanol:H2O (1:1 v/v) 90 ml
glacial acetic acid 10 ml
Filter the solution before use.
Store at room temperature for a few months
2. Protein A/G plus Agarose Santa Cruz Biotechnology (cat. n. sc 2003)
3. Glacial acetic acid Prolabo (cat. n. 20103.320)
4. Comassie brilliant blue Sigma (cat. n. B-0149)
1. Ultracentrifuge
2. Sambrook, J., E.F. Fritsch, T. Maniatis. 1989. Molecular cloning, a laboratory manual. Cold Spring Harbor Laboratory, eds.
3. Ridley, A.J., and A. Hall. 1992. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 70: 389-399.
4. Immunocytochemical technology. G. V. Childs, ed. Alan R. Liss, Inc., New York.
5. Harlow, E., D. Lane. 1988. Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory, eds.
6. Coons, A.H., and M.H. Kaplan. 1950. Localization of antigens in tissue cells. Improvements in a method for the detection of antigen by means of fluorescent antibody. Journal Experimental Medicine 91: 1-13.
7. Wary, K.K, F. Maniero, S.J. Isakoff, E.E. Marcantonio, and F.G. Giancotti. 1996. The adaptor protein shc couples a class of integrins to the control of cell cycle progression. Cell 87 (4): 733-742.
8. Defilippi, P., M. Venturino, D. Gulino, A. Duperray, P. Boquet, C. Fiorentini, G. Volpe, M. Palmieri, L. Silengo, and G. Tarone. 1997. Dissection of pathways implicated in integrin-mediated actin cytoskeleton assembly. J. Biol. Chem. 272: 21726-21734.
9. Rouslahti, E. 1997. Stretching is good for a cell. Science 276, 1345-1346.
10. Fiorentini, C., A. Fabbri, P. Matarrese, L. Falzano, P. Boquet, and W. Malorni. 1997. Hinderance of apoptosis and phagocytic behaviour induced by E. coli cytotoxic necrotizing factor 1 (CNF1): two related activities in epithelial cells. Biophys. Biochem. Res. Com. 241:341-346.
11. Haldar, S., A. Basu, and C.M. Croce. 1997. Bcl2 is the guardian of microtubule integrity. Cancer Res. 15: 229-233.
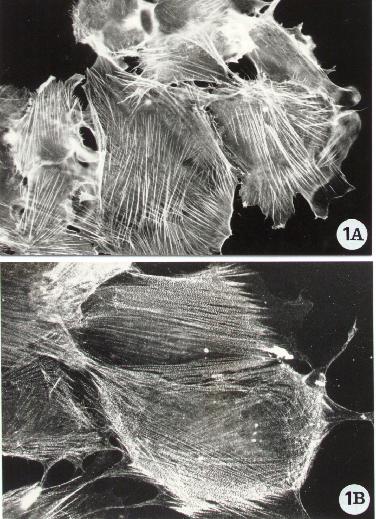
Fluorescence microscopy of human epithelial cells stained
for the detection of F-actin (A) and of the actin-binding protein alpha-actinin
(B). Following the procedure reported in Methods A2.2., F-actin was directly
evidenced with FITC-phalloidin whereas alpha-actinin by an indirect method,
standard protocol, using a specific monoclonal antibody (Chemicon). To
note F-actin organized in stress fibers and alpha-actinin distributed along
the actin filaments.
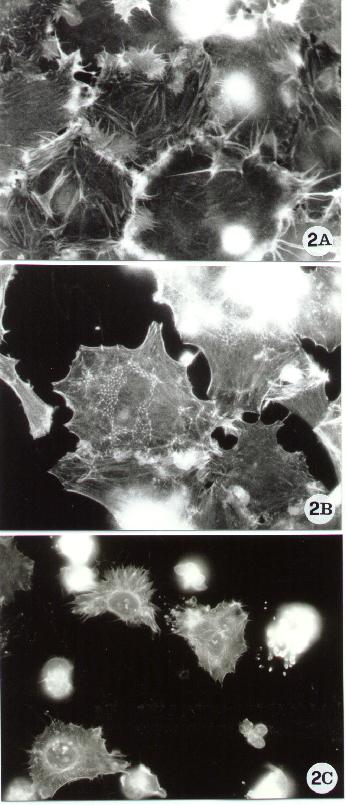
Figure 2.
Fluorescence microscopy of human epithelial cells stained with FITC-phalloidin for F-actin detection, as reported in Methods A.2.2.. The figure shows some of the actin cytoskeletal alterations which have been associated with apoptosis. Spreading cells, characterized by membrane ruffling (A) and by a peculiar thin meshwork (B) are known to protect cells against apoptosis (9). By contrast, apoptosis often causes the rounding up and the breakdown of the actin network (10).
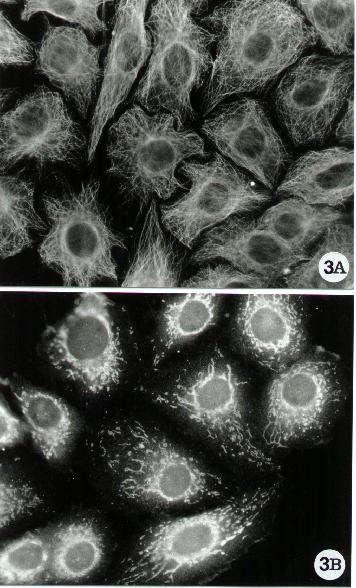
Figure 3.
Fluorescence microscopy of human epithelial cells stained
for microtubular network (A) and Bcl-XL detection (B). Bcl-XL is an anti-apoptotic
protein of the Bcl-2 family, associated with microtubules (11). In both
cases, it was used the indirect method reported in the standard protocol,
Methods A.2.2. Tubulin was evidenced by using a mixture (1/1) of monoclonal
anti a and b tubulin
(Sigma) and Bcl-XL with a specific anti rabbit polyclonal antibody (Santa
Cruz). To note the distribution of the anti-apoptotic protein which overlaps
the microtubular network.
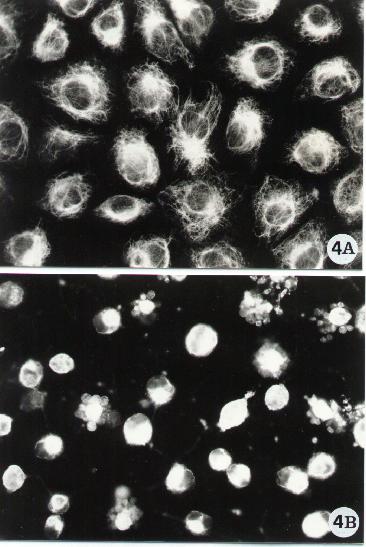
Figure 4.
Fluorescence microscopy of human epithelial cells stained for the detection of the intermediate filament-type vimentin. Vimentin was evidenced by a monoclonal antibody (Sigma) following the indirect method described in Methods A.2.2. To note the arrangement of vimentin in control cells (A) and the breakdown of its network (B) upon exposure to an apoptotic inducer (in the micrograph is shown the effect of a bacterial toxin known to cause apoptosis in epithelial cells).
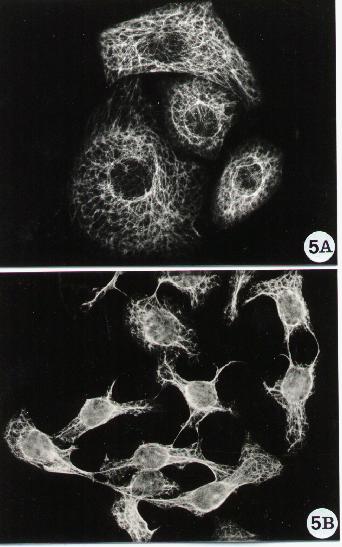
Figure 5.
Fluorescence microscopy of human epithelial cells stained
for cytokeratins' detection. A monoclonal antibody anti-pan-cytokeratin
(Sigma) was adopted in the indirect method described in Methods A.2.2.
To note that the cytokeratin network in epithelial cells (A) can undergo
collapse when treated with apoptotic agents which influence the cell shape
(B).